Light–matter interactions: Lasers, laser spectroscopy, and photodynamics
Of all the phenomena studied by physical chemists, perhaps none has been more centrally important than light–matter interactions. There are many techniques by which physical chemists have interrogated matter. Nothing has taught us more and has more to teach us than light–matter interactions in the form of spectroscopy and photochemistry. It is the discovery of lasers and the application of lasers that has opened up the world of how atoms and molecules interact more than any other technique. And it is the application of new laser techniques, including microspectroscopy and spectromicroscopy, that has more to tell us about intermolecular and interatomic interactions than any other technique. Of course, lasers are not the only word in spectroscopy and microscopy. Nuclear magnetic resonance (NMR) forms the foundation of structural analysis along with diffraction techniques. These techniques are also being pushed ever more into the time-resolved domain and dynamical studies. Electron microscopy is becoming more relevant to chemistry, and mass spectrometry is essential in the chemist’s arsenal. This chapter, however, will focus on lasers and their interactions with the frontiers of chemistry, physics, and biology.
27.1 Lasers
The introduction of lasers is one of several groundbreaking technologies that has transformed physical chemistry. The others are molecular beams, routine attainment of (ultra)high vacuum and high-speed electronics (along with the accompanying detectors and computers). The last two have also facilitated advances in electron microscopy, mass spectrometry and NMR. Lasers are transformative to research because they combine a number of exceptional traits:
- Monochromaticity. Most lasers operate at a wavelength with a well-defined central value and a small bandwidth about this central value.
- Directionality. Lasers naturally form beams with low divergence. This means that they are easily directed with conventional optics such as mirrors and can be tightly focused with lenses. Just try this with a light bulb.
- Pulsed versus continuous-wave operation. Lasers can be operated in a continuous-wave (CW) or pulsed mode, based on their construction without the need for mechanical modulation. CW operation is required for the highest monochromaticity. Pulse widths of lasers can range from microseconds to femtoseconds. Lasers can also be used to generate attosecond pulses.
- Brightness and intensity. Combining the first two properties, lasers deliver extremely high values of photons per unit bandwidth per unit area. When crammed into extremely short pulses this corresponds to extremely high energy density (J m−2), power density (J s−1 m−2) and electric field strengths (even exceeding the electric field that binds electrons to nuclei).
- Coherence. The photons in a laser beam are in phase. This happens both transversely and longitudinally (axially). That is, there is a well-defined phase relationship across the diameter of the beam as well as along the propagation direction. Another way to say this is that the coherence is both temporal and spatial.
Infrared and visible lasers were inspired by the work of Arthur Schawlow and Charles Townes.1 The first laser was a ruby laser with millisecond pulses. This was demonstrated in 1960 by Theodore Maiman at the Hughes Research Laboratories. In the press conference announcing the discovery, Maiman said that the laser was ‘a solution seeking a problem.’2 It would not take long for chemists and physicists – who are never short of problems – to start matchmaking between lasers and problems. The first CW laser followed the next year. This was the HeNe laser.3 Such a laser was used in the first laser-induced fluorescence experiments,4 which was one of its first applications to chemistry. The dye laser invented by Mary Spaeth at the Hughes Aircraft Company in 1966 was followed independently and with improvements by Peter P. Sorokin at IBM and Fritz P. Schäfer at the Max-Planck-Institut für biophysikalische Chemie. The dye laser offered tunability, which was essential for its applications to chemical physics. The age of lasers was now set to take off.
The operation of a laser depends on the recognition that there are three ways to link two levels radiatively. The absorption of light is stimulated by the presence of on-resonance photons. The emission of light is either spontaneous, initiated by fluctuations in the vacuum field that fills the universe, or stimulated, initiated by the presence of on-resonance photons that we add directly to the system. The net rate of stimulated emission depends on the population difference between the upper and lower states.

Normally (in thermal equilibrium) more population is in the lower energy state. When more population is in an excited state compared to a lower state, we call this a population inversion. A population inversion is one basic requirement for making a laser (unless something exotic is going on). Consider a two-level system with total population N and the population of the two individual states given by Ni and Nk,

The population difference is

Population of the two levels is changed by the absorption and emission of photons. The rate of change of the population difference is

If the energy difference between states is large such that we can neglect thermal population of the excited state, then initially ΔN = N. Thus

and the population difference is being driven down because irradiation drives transitions between the two levels. At steady state dΔN/dt = 0 and

In the limit of a large driving field (strong irradiation), this is driven to saturation at ΔN = 0. At saturation there is no net emission of photons. A two-level system can never lase as it kills its own inversion. A three- or four-level system must be used so that a population inversion can be sustained.
This idea of saturation has important implications beyond the construction of lasers. It means that once we have constructed a laser, we cannot use a photon field (no matter how intense) to move all the population from the ground state to the excited state of a two-level system. Once 50% of the population has been moved to the excited state, the probability that the laser beam excites more molecules to the excited state is the same as the probability that the laser beam stimulates the emission of photons and moves population back to the ground state. There is only so hard we can pump a transition before we start to pump population down as well as up. At this point applying more intensity will not change the net rate of absorption (which is zero); however, it may change the rate of other processes that previously were low-probability events (such as two-photon absorption).
Let us also plant a fertile idea that is rooted in stimulated emission and saturation. We see from this discussion that is possible not only to move population upwards by stimulated absorption, but also to move population downward by stimulated emission. Furthermore, if we introduce two different lasers with two different colors (which is therefore called a two-color experiment), the stimulated processes move population between two different sets of states.
The production of lasing action depends on pumping a population inversion in a three- or four-level system. The ruby laser is a three-level system. The four-level system of the much more important and popular Nd:YAG laser (often simply called a YAG, which is used to make the green beam in Fig. 27.1(a)) is illustrated in Fig. 27.1(b). A population inversion is required so that stimulated emission (gain) occurs at a higher rate than absorption (loss).

Figure 27.1 (a) A Nd3+:YAG-pumped femtosecond dye laser. (b) The Nd3+:YAG system is an example of a four-level laser system. When sufficient pump power Wp is applied a population inversion between levels |3⟩ and |4⟩ is established and lasing can occur. Fast nonradiative processes (dashed lines) move population into the upper state and remove it from the lower state to maintain the population inversion.
Pumping requires an energy source that can be directed into the optical medium. Just about anything can be used for this. The best source is a laser – which is, of course, difficult if you are trying to invent the first laser! Excitation can be provided by a discharge, particularly for gas lasers, or simply as current for semiconductor/diode lasers. Flashlamps for pulsed operation or arc lamps for CW operation can be used for optical excitation. A select few chemical reactions are able to produce a population inversion directly that can be used as the optical medium. A nuclear blast provides copious energy and has been used for the pumping of high-energy (X-ray) transitions. But nuclear bombs tend not to be very useful in a laboratory setting. The power to drive a laser scales as the fifth power of the frequency of the laser, Ppump∝v5. This effectively prevents conventional laser schemes from extending far beyond the UV.
The strategy for obtaining a population inversion is illustrated in Fig. 27.1(b). A pump source excites the Nd3+ ion. A flashlamp is a simple but inefficient means of pumping, as most of the light is not resonant with the pump band. Extremely efficient pumping with much less heat generation is performed by a diode laser tuned to 810 nm to overlap the pump band resonantly. Nd3+ is an ion with electron configuration 4f3. It is doped into crystalline Y3Al5O12 (yttrium aluminum garnet or YAG) at a level of 0.5–2.0 wt%. Its electronic levels are sharp and do not interact strongly with the lattice. It is not tunable, but it can be operated either in a pulsed or CW mode. The excitation is strong and moves a lot of population to the pump levels. A fast transfer of population occurs from this set of levels to the upper state of the transition that will be involved in lasing. This 4F3/2 state has a forbidden transition with the 4I11/2 state. Importantly, transition to the ground state is even more forbidden; this means that population can build up in the 4F3/2 state rather than being lost to spontaneous emission.
Eventually, spontaneous emission lets loose a photon on the 4F3/2 → 4I11/2 transition. This photon encounters a Nd3+ ion that has been excited to the 4F3/2 state. The photon is resonant with the transition to the 4I11/2 state, and therefore it stimulates the emission of a second photon. Not only does it stimulate emission of a photon with the same wavelength, but the emitted photon has the same phase and the same direction of propagation as the first photon. These two coherent photons propagate and stimulate more emission. As long as there is more population in the upper state than the lower state, the number of new photons created by stimulated emission will outnumber the number of photons consumed by absorption. The result is a monochromatic, coherent, directional beam of light. The final requirement of the four-level system is that the lower level of the lasing transition is strongly coupled to the ground state so that the population inversion is easier to maintain.
The most efficient way to build a laser is to start with a laser. The photograph in Fig. 27.1(a) is of a Nd3+:YAG-pumped femtosecond pulsed dye laser. The YAG laser pumps both the oscillator and amplifier of the dye laser. The oscillator and the amplifier are two different optical elements that are important for the construction of lasers. Any region of space in which a population inversion exists can act as an amplifier. An amplifier is usually used in a single-pass or few-pass mode. The medium is pumped to obtain a population inversion. Light that enters the amplifier interacts with the population inversion to stimulate emission. The output intensity is amplified by some factor determined by the extent of the population inversion and the path length. Some lasers operate in this manner; for example, excimer lasers and CO2 lasers are sometimes constructed of only an amplifier. An amplifier does not lead to particularly good beam quality in terms of divergence and coherence, especially if they rely on a single pass through the medium.
An oscillator is required to get the best beam quality. An oscillator, as the name implies, allows the light to circulate many times back and forth between highly reflective mirrors. This creates a beam with superior qualities of monochromaticity, directionality, brightness and coherence. In an oscillator, only those modes that fit exactly inside the cavity will be amplified. Only light for which an integer number of half-wavelengths n matches the cavity length d fulfills the resonance condition,

Most lasers are run in multimode operation. The highest degree of monochromaticity requires single-mode operation, which will also generally improve the coherence and the uniformity of the intensity profile of the laser. It also comes with the cost of considerably reduced intensity.
If the oscillator mirrors are highly reflective, very little of the laser light escapes from the oscillator. While this provides for many round-trips in the oscillator, it also means that the optical power circulating in the optical cavity is possibly orders of magnitude higher than what is escaping out of the end mirror (also called an output coupler). Such an oscillator is said to have a high quality factor or high Q factor. If we could suddenly switch one mirror from begin 100% reflective to 0% reflective – that is, if we could switch the oscillator from being a high-quality optical cavity to a low-quality optical cavity – then we could let a pulse of laser light out of the oscillator. This is known as Q switching, and can be used to generate nanosecond pulses with repetition rates from single pulses to the kHz range. Often, this is done with polarization tricks and acousto-optic or electro-optic modulators. For instance, the mirror might be 100% reflective for vertical polarization of light but 0% reflective for horizontal polarization. By using the modulator to switch back and forth between the two linear polarizations, pulses can be coupled out of the oscillator as a regular pulse train.
An example of a laser that can be run in single-mode operation is the Ti:sapphire laser, which crucially is tunable in the range 670–1100 nm. Crucial because this extremely high-gain, broad bandwidth facilitates the production of pulses as short at 5 fs. Sapphire is crystalline Al2O3. This is doped with around 0.1 wt% Ti2O3. Oscillator and amplifier crystals are doped to different densities. The Ti3+ ion has the electron configuration 3s2 3p6 3d1. The 3d orbitals are crystal field split into a triply degenerate lower-energy T2 orbital and an upper doubly degenerate E orbital. The ground state is 2T2 with a 2E excited state. These levels are separated by ≈19 000 cm−1.
Figure 27.2(b) demonstrates how, even though only two electronic states are involved in the laser transition, the system still effectively acts as a four-level system. The upper state is split by the Jahn–Teller effect. The upper band can be pumped in the blue, but it is more efficient to pump the lower energy component in the green. Only the lower band is shown in Fig. 27.2(b). The Jahn–Teller distortion in the 2E state leads to intrinsic coupling of the state to the lattice and a large shift in the Franck–Condon region for absorption from the excited state potential minimum. Relaxation in the excited state is an ultrafast process that occurs much more rapidly than radiative relaxation to the ground state. Population builds up at the minimum of the excited state, which then relaxes radiatively at wavelengths greatly red-shifted compared to the pump. A broad Franck–Condon region for this transition leads to emission over a broad wavelength range. Efficient vibrational relaxation in the ground state removes population from the lower level of the lasing transition and returns it to the ground state. Because of the essential role of lattice coupling and vibrational relaxation, the Ti:sapphire laser is sometimes called a vibronic laser.
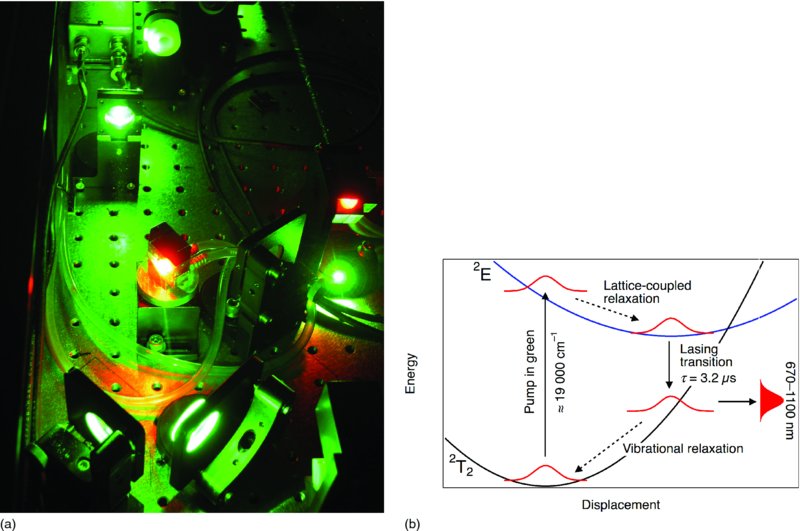
Figure 27.2 (a) A diode-laser-pumped regeneratively amplified Ti:sapphire laser system. The green light is from the pump laser. Two Ti-doped sapphire crystals fluoresce red and act as two stages of amplification. The oscillator is a separate laser in this system. (b) The level scheme of the Ti:sapphire laser.
The Ti:sapphire laser can operate in either CW or pulsed mode. Mode locking is essential for operation in the femtosecond regime. Although normally it is not a good idea to hit your laser, it was discovered accidentally that vibration of the sapphire rod, which can be caused by striking the table on which the laser sits, can force the Ti:sapphire system to self-mode lock. Piezoelectric modulators are used to perform this task controllably. A broad range of laser systems are described in Table 27.1.
Table 27.1 A survey of laser systems and their characteristics.
| Laser material | λ/nm | hν/eV | Characteristics |
|---|---|---|---|
| Solid state | |||
| Semiconductor laser diode | IR–visible ~0.4–20 μm | Usually CW but can be pulsed; wavelength depends on material; GaN for short λ, AlGaAs 630–900 nm, InGaAsP 1000–2100 nm, used in telecommunications, optical discs | |
| Nd3+:YAG (1st harmonic) Nd3+:YAG (2nd harmonic) Nd3+:YAG (3rd harmonic) Nd3+:YAG (4th harmonic) | 1064 532 355 266 | 1.16 2.33 3.49 4.66 | CW or pulsed, ~10 ns pulses, most common, 150 ps versions (and shorter) available, 10–50 Hz rep rate, 1 J to many J pulse energies. Nd3+ can also be put in other crystalline media such as YLF (1047 and 1053 nm) or YVO4 (1064 nm) |
| Nd3+:glass | 1062 or 1054 | 2.33 | ~10 ps, can be used to make terawatt systems for inertial confinement fusion studies |
| Ruby (Cr:Al2O3 in sapphire) | 694 | 1.79 | ~10 ns pulses when Q switched, first laser system |
| Ti:sapphire | 700–1000 | 1.77–1.24 | fs to CW; from low repetition rate to 82 MHz |
| alexandrite (Cr3+ doped BeAl2O4) | 700–820 | 1.77–1.51 | tattoo removal |
| Liquid | |||
| dye laser | 300–1000 | 4.13–1.24 | rep rate and pulse length depend on pump laser; fs, ps, ns up to CW |
| Gas | |||
| CO2 | 10600(10.6 μm) | 0.12 | Long (many μs), irregular pulses, CW or pulsed at high rep rates, line tunable, few W to >1 kW |
| Kr ion | 647 | 1.92 | CW, line tunable, 0.1–100 W |
| HeNe | 632.8543.5 | 1.962.28 | CW, 0.5–35 W |
| Ar ion | 514.5488 | 2.412.54 | CW, line tunable |
| HeCd | 441.6, 325 | 2.81,3.82 | CW, 1–100 mW |
| ArF excimer | 193 | 6.42 | ~20 ns, 1– >1000 Hz, several W to over 1 kW, 100 mJ to >1 J |
| KrF excimer | 248 | 5.00 | 30–34 ns |
| XeCl excimer | 308 | 4.02 | 22–29 ns |
| XeF excimer | 351 | 3.53 | 12 ns |
| F2 | 154 | 8.05 | 1– several kHz rep rate, 1–20 W, 10–50 mJ pulse energies, 10 ns |
| N2 | 337 | 3.68 | 1–3.5 ns, 0.1–1+ mJ, 1–20 Hz rep rate |
| Exotic | |||
| Free electron laser | 1 nm–THz | Wide variety of characteristics depending of machine characteristics | |
| X-ray lasers | Lasing medium is a highly ionized plasma producing light at one wavelength | ||
| High harmonic generation | UV–X-ray | High-powered laser pumps a gas to produce light at wavelengths that are harmonics of the pump wavelength. Capable of producing attosecond pulses. |
27.1.1 Directed practice
For a Nd3+:YAG laser operating on its fundamental wavelength in a 10 cm cavity, calculate the wavelength spacing between two successive modes.
[Answer: 5.66 pm]
27.2 Harmonic generation (SHG and SFG)
An incident electromagnetic wave induces a polarization in the medium it strikes given by

where ϵ0 is the vacuum permittivity, χ(k) is the kth order nonlinear susceptibility of the medium, and E is the electric field vector. Any phenomenon that depends on one of the k ≥ 2 terms is called a nonlinear effect because it depends on the field strength raised to a power larger than one. In the multipole expansion of the nonlinear susceptibility χ(k) each term of successively higher order is much smaller than the previous term. On symmetry grounds some terms vanish. For instance, in any centrosymmetric medium (a medium that contains an inversion center such as an elemental crystal or isotropic material) all even order terms are zero.
The linear term relates to the oscillating electric field with amplitude A and angular frequency ω,

The second-order term oscillates according to

Thus, radiation scattered as a result of the χ(2) term oscillates at twice the incident frequency. It has been frequency-doubled. Equivalently, this second harmonic signal is generated at half the wavelength of the incident light. The intensity in the second harmonic signal increases with the square of the incident intensity. Effectively, the induced polarization field takes two photons from the incident field, adds them together and emits them as one photon of twice the energy.
Second harmonic generation (SHG) does not happen in a gas or a glass. There are a number of crystalline materials that are particularly effective at second harmonic generation that find widespread use in nonlinear optics. These include potassium dihydrogen phosphate (KDP), β-barium borate (β-Ba2O4) and lithium niobate (LiNbO4). These crystals are often inserted into laser beams to generate light at frequencies that cannot be generated directly by the oscillator or amplifier. SHG is also possible at the interface of two materials. In this way, SHG can be used to probe molecules adsorbed at the interface. If a second laser is incident on the polarization induced by the first laser, the χ(2) term of the material will, with a probability equal to the product of the intensity of the two lasers, generate a response at the sum frequency. In other words, the χ(2) term can add two photons of different frequencies to form one photon with an energy equal to the sum of the two photon energies. This process is called sum frequency generation (SFG). Both, SHG and SFG are much more likely to occur if one of the frequencies involved in the process is resonant with a molecular process. For example, a common application of SFG is to perform time-resolved vibrational spectroscopy.
Since its introduction by Ron Shen,5 a very common combination is to choose an infrared frequency, which can be tuned through vibrational resonances of an adsorbate, and to mix it with a visible or near-IR photon of fixed frequency to yield a visible photon. The fixed-frequency pulse is called the up-conversion pulse. In time-resolved studies, the tunable pulse is used to pump population into an excited state, and is called the pump pulse. The fixed-frequency pulse (probe pulse) then upconverts the polarization field at the frequency of the first pulse into the photon that is detected. The signal intensity in such a pump–probe experiment is determined not only by the intensities of the pump and probe pulses, but also by the lifetime of the excited state and the delay between the two pulses, which allows for direct determination of the lifetime of the excited state. The delay between the two pulses, as shown in Fig. 27.4(a) can be controlled by changing the position of a mirror, which introduces a controlled path length difference between the pump and probe beams.

Figure 27.3 A semiconductor diode laser. (a) In the unbiased state there is no overlap between electron and hole state so there is no radiative recombination. (b) When biased, hole states in the valence band of the p-doped region become available for recombination with electrons from the n-doped region.

Figure 27.4 (a) Second harmonic generation (SHG) is illustrated. Two coherent beams, created by splitting the beam generated by one short-pulsed laser, are focused on the same spot in a sample. (b) The spectrum of the SHG beam, centered at 514 nm for 1028 nm incident wavelength, reflects the bandwidth of the incident light. (c) By varying the spatial and temporal overlap of the beams the signal is optimized. For nonresonant SHG, the cross-correlation signal in panel (c) is a measure of the pulse width. For resonant SHG through a real state with a finite lifetime, the cross-correlation signal is now a convolution of the pulse width and the intermediate state lifetime. (d) In sum frequency generation (SFG) two different wavelengths, visible (vis) and infrared (IR) in this example, are mixed on the sample. Panels (a–c): Images provided by Sylvie Roke and reproduced with permission from Macias-Romero, C., Didier, M.E., Jourdain, P., Marquet, P., Magistretti, P., Tarun, O.B., Zubkovs, V., Radenovic, A., and Roke, S. (2014) Opt. Express, 22, 31102; © Optical Society of America. Panel (d): Roke, S. and Gonella, G. (2012) Annu. Rev. Phys. Chem., 63, 353. © 2012, Annual Reviews.
27.2.1 Directed practice
Calculate the wavelength of the third harmonic of a Ti:sapphire laser operating at 800 nm. Calculate the sum-frequency photon created by mixing a 532 nm photon with a photon resonant with a vibration at 2100 cm−1.
[Answers: 267 nm, 685 nm]
27.3 Multiphoton absorption spectroscopy
SHG, SFG and the Raman effect are all nonlinear spectroscopies. The signal level in nonlinear spectroscopies usually changes as In, where I is the laser intensity and n is an integer greater than one that often equals the number of photons in the process. There is a much broader branch of spectroscopy involving transitions mediated by more than one photon. The theory of multiphoton transitions in atoms was founded by Maria Goeppert-Meyer in her doctoral thesis. This was before the invention of the laser. Therefore, experimental verification of any of this theory appeared extremely unlikely at the time she submitted her thesis. Faced with this, she changed the focus of her research. And a good choice it was, because she was to become the first woman since Marie Curie in 1903 to be awarded the Nobel Prize in Physics. Goeppert-Meyer received hers in 1963 for her discoveries pertaining to nuclear shell structure.
The understanding of multiphoton transitions is greatly simplified with the following conceptual framework. Solutions of the Schrödinger equation for a molecule reveal the wavefunctions that describe the states of the molecule – the molecule in the absence of a photon field. When applied at a high enough intensity, states of the molecule + field system emerge. We call these molecule + field states virtual states. Another way to put it is that virtual states are states of the molecule that only exist in the presence of the photon field. Virtual states are available for transitions just as are real states. One interesting property of virtual states is that they draw their existence and properties from the real states of the molecule. In particular, they draw the largest contributions from the states nearest to them. The strength of transitions through virtual states differs from one state to the next, just as it does for transitions between real states. The more character a virtual state can draw from real states, the stronger transitions through it will be.
The selection rules for two-photon absorption are the same as for vibrational Raman transitions. The cross-product of the symmetry species that characterize the two electronic states and the two-photon tensor S must contain a totally symmetric character,

The two-photon tensor S transforms as does the polarizability tensor  , that is,
, that is,

This is particularly important to recognize for determining two-photon transition selection rules in polyatomics, and whether the transitions are vibronically allowed. For multiphoton transitions of nth order, one can also apply a ‘chain rule’ to determine if multiphoton transitions are allowed in which the one-photon selection rules are applied for each ‘link’ in the multiphoton process. This is particularly simple for diatomics, which have a simple set of selection rules for one-photon transitions.
- First, spin conservation. The spin selection rule is the same for any nth order transition: the spin quantum number does not change, either ΔS = 0 or ΔΣ = 0 as appropriate.
- Next, orbital angular momentum conservation. ΔΛ = 0, ±1 for each photon involved in the multiphoton process. Thus, for a two-photon process, ΔΛ = 0, ±1, ±2 result from all combinations of ΔΛ = 0, ±1 taken twice. In a three-photon process, ΔΛ = 0, ±1, ±2, ±3 are allowed.
- Next, parity conservation. In a one-photon Σ–Σ transition, the parity must not change. Since one photon cannot change parity, so too n photons cannot change parity. The parity does not change regardless of the number of photons involved (+ remains + or – remains –).
- Finally, consideration of inversion symmetry. Inversion symmetry must change in any one- or odd-photon transition (g or u must change). Inversion symmetry must not change in any even-photon transition (g or u must stay the same).
27.3.1 Laser-induced fluorescence (LIF)
In multiphoton spectroscopy we have to keep track of whether we are adding photons independently or coherently, as well as the bandwidth of the light source. Conventional fluorescence involves two independent photons. The first photon excites a molecule to an electronic excited state. A second photon is then emitted independently by spontaneous emission, as shown in Fig. 27.5(a). The fluorescence can be detected in one of two modes: (i) we can detect all of the light without dispersion of the wavelength (total fluorescence); or (ii) we can measure the spectrum of the emitted light by dispersing it with, for example, a grating (dispersed fluorescence). What we usually call a fluorescence spectrum is more precisely called a dispersed fluorescence spectrum with constant excitation wavelength. In this case, the spectrum is a plot of fluorescence intensity as a function of the fluorescence photon wavelength, as shown in Fig. 26.13 for rhodamine 6G (R6G). It probes how the excited state relaxes to a lower state.

Figure 27.5 Laser-induced fluorescence (LIF). A laser is used to excite the system from state 1 to state 3 by either one-photon absorption, as in process (i), or two-photon absorption, as in process (ii). Subsequently, spontaneous emission (fluorescence) is detected between the excited state 3 and lower level 2.
We can also acquire an excitation spectrum. In one type of excitation spectrum, we disperse the fluorescence and detect the light emitted at one wavelength while changing the excitation wavelength. Alternatively, we collect the undispersed light while scanning the excitation wavelength. In either case, the spectrum obtained is a plot of fluorescence intensity versus excitation wavelength, and the spectrum resembles an absorption spectrum.
Conventional fluorescence spectroscopy utilizes a broadband source to excite a sample. A spectrometer disperses the light onto a detector. This technique obtains high resolution at the expense of signal intensity.6 With the use of narrow bandwidth and intense laser excitation, we can perform high-resolution spectroscopy based on the absorption of photons that is also highly sensitive. A laser with a bandwidth smaller than the spacing between spectral lines excites the molecule quantum-state specifically. The excited state population is directly proportional to the initial state population. The fluorescence signal is then proportional to the initial state population. In this manner a spectrum of the initial state molecule is acquired by detecting either the dispersed or undispersed fluorescence emitted when the laser is tuned across the frequency range in which the initial states absorb light. If the electronic transition is excited to saturation, we move the maximum amount of population into the excited state. At this point it can be shown that the ratio of the intensity of two transitions in the same rotational band and the same band progression (i.e., Δv constant) Ii/Ij is directly proportional to the population ratio of the two initial states ni/nj,

In the absence of saturation, the line strength of the transition (its Einstein B coefficient) and the laser power (in terms of the energy density) must also be taken into account according to

This technique is known as laser-induced fluorescence. It is a sensitive method for the measurement of quantum state distributions, and was introduced by Richard N. Zare.7 First performed with CW lasers such as the HeNe and Ar ion laser, its applications skyrocketed with the introduction of tunable lasers such as the dye laser. When applied to atoms it can resolve spin–orbit splittings between different J states in a given electronic level. After excitation, the undispersed fluorescence is collected to obtain the highest detection efficiency. High detection efficiency is achieved because we know when to look for a signal (after the laser pulse has excited the sample), and because we detect against zero background. A background of nothing but electronic noise is achieved because the time delay between excitation and detection allows us to wait for scattered laser light to be absorbed, and because the fluorescence is red-shifted compared to the excitation light to further enhance its separation. Single-molecule detection has been possible with LIF.
LIF is normally used to measure the population in the initial state after it has been populated by scattering, a chemical reaction, or expansion in a molecular beam. This is particularly true when undispersed fluorescence is acquired as a function of the excitation laser wavelength. Dispersing the fluorescence allows us to probe the transitions from a quantum state selected by the excitation laser to a lower electronic level. Thereby, we can probe the relaxation dynamics of the upper state. The fluorescence can either be collected by lenses and mirrors onto a photomultiplier, or else it can be imaged to obtain more detailed information about spatial distributions. The use of pulsed lasers also allows us to introduce delays and time resolution into our data analysis. Great sensitivity and the ability to produce detectable signal in the few nanoseconds of a pulsed laser also facilitates the detection of transient species, such as reactive intermediates produced in chemical reactions, even combustion. LIF is capable of detecting nascent reaction products – that is, reaction products with quantum state distributions as produced by the reaction and before they have collided with any other species. The polarization of the incident light and/or the fluorescence can also be analyzed. This allows us to determine whether the sample is aligned or oriented following a reaction8 or scattering.9 An aligned sample is one that rotates in a preferred plane. An oriented sample is one that not only rotates in a preferred plane but also has a preferred sense of rotation – that is, clockwise at opposed to counterclockwise.
An LIF signal can be collected from essentially any volume into which a laser can be focused. Thus, it can be used not only to detect molecules in a gas cell but also inside of a modified diesel engine to study combustion reactions, or in a molecular beam to measure the rotational temperature of expanding molecules. By translating the focal volume, spectroscopic information can be obtained with spatial resolution. As we will discuss below, by using more than one laser beam we can even beat the diffraction limit in spatial resolution. LIF also works in liquids. One area that has been particularly fruitful in this respect is the detection of molecules after they have been separated by capillary electrophoresis (CE). Indeed, LIF detection of CE effluent was used to readout the millions of bases in DNA required to sequence the human genome for the first time.
Laser excitation to induce fluorescence is not limited to one-photon processes. Coherent absorption of two or more photons can also be used to excite fluorescence. This is exploited in two-photon fluorescence microscopy, most commonly with a femtosecond-pulsed Ti:sapphire laser as the excitation source.
27.3.2 Stimulated emission pumping
Laser-induced fluorescence is most commonly implemented as an incoherent two-photon technique in which the first photon comes from a laser and the second photon is emitted from the molecule spontaneously and randomly based on the lifetime of the excited state that was created by absorption of the laser photon. If we introduce a second laser that is resonant with a transition from the excited state created by the absorption of the first photon to a lower energy state, then we do not need to wait for spontaneous emission. Instead, we force the system to relax. Moreover, we can determine the state to which the photon relaxes by tuning the wavelength of the photon to be resonant with a particular transition. This process of pumping population into an excited state with one laser, then dumping population into another (but lower energy) excited state with a second laser, is known as stimulated emission pumping (SEP) and is illustrated in Fig. 27.6. SEP has three important applications: (i) it selectively moves population to a specific quantum state; (ii) it facilitates pump-dump-and-probe detection; and (iii) it selectively turns off fluorescence in the region of space depleted of excited state population by the dump laser. The excited state does not have to be a real state. If the excited state is a virtual state instead of a real state, the technique is known as stimulated Raman scattering (SRS). In either case (SEP or SRS), the difference in energy between the pump and dump photons matches the energy difference between the initial and final molecular states. SRS has the disadvantage that practical schemes can only change the vibrational quantum number by 0 or ±1. SEP can, in principle, be used to change the vibrational quantum number by an arbitrary amount, limited only by Franck–Condon factors to determine the efficiency. However, appropriately tunable lasers must be found in either case, which can be problematic depending on the molecule under consideration.

Figure 27.6 Stimulated emission pumping (SEP) of state 4 occurs by populating state 3 with the absorption of photon λ1, then stimulating the transition 3 → 4 by resonant irradiation at wavelength λ3. Population of state 4 can be followed by, for example, detecting its fluorescence at λ4. Stimulated Raman scattering (SRS) can also be used to pump levels to quantum state specifically. Regardless of whether pumping is performed resonantly, for example using photons with wavelengths λ1 and λ2, or nonresonantly through a virtual state, using photons with wavelengths λ5 and λ6, the difference in photon energy must be resonant with the difference in energy between the initial state and the state pumped.
The first application of SEP is the preparation of a specific quantum state. Selective population of a quantum state allows us to study the behavior of a reactive system in a quantum-state-specific manner. For instance, suppose you would like to study how the rotational and vibrational states of a molecule change when, for instance H2 or NO is scattered from a metal or insulator surface,10 or to test whether the Born–Oppenheimer approximation is appropriate for describing these interactions and reactions on surfaces.11 Then either SRS or SEP can be used to transfer population to specific quantum states in a molecular beam. The molecular beam is aimed at a surface, and the quantum state distribution before and after interaction with the surface can be measured using resonance-enhanced multiphoton ionization (REMPI; see below).
The second application is a pump, dump, and probe experiment. This is a three-color experiment in that three different photons are used. One photon prepares an excited state (pump), while the second photon moves population to a different quantum state by stimulated emission pumping (dump). The third or probe photon ionizes the molecule (or induces LIF) so that the ions can be detected and quantified. The use of three separate photons allows us to potentially introduce two variable delay times. Suppose, for instance, you want to study the dependence of the ionization rate of I2 in a strong laser field as a function of the internuclear distance R.12 You could pump the molecule to an excited state by a Franck–Condon transition. Since you know that the molecule is excited onto the repulsive wall of the excited state potential, you know that the molecule’s bond will stretch after excitation. By waiting a known amount of time after the pump pulse, you can control how much the internuclear separation increases before you dump the molecule back to the ground electronic state. Subsequently, the third photon probes the ground state by ionization and the ionization rate can be measured as a function of R. Similarly, the reactivity of a prepared state can be measured by delaying the time between the pump, dump and probe pulses.
The third application is detailed in the next section.
27.3.3 Superresolution fluorescence microscopy
In Chapter 19 we mentioned that the ultimate resolution of a microscope Δmin is given by the Abbe limit, and is roughly Δmin ≈ λ/2. A more refined derivation13 of this limit demonstrates that for a point source propagating along the z-axis, the resolution deviates slightly from this formula. More importantly, the optimum resolution is significantly worse in the z-coordinate than in the lateral xy-plane. In the xy-plane the ultimate lateral resolution is given by

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


