FIGURE 7-1 Lidocaine serum concentrations initially drop rapidly after an intravenous bolus as drug distributes from blood into the tissues during the distribution phase. During the distribution phase, drug leaves the blood due to tissue distribution and elimination. After 0.5-1 hour, an equilibrium is established between the blood and tissues, and serum concentrations drop more slowly since elimination is the primary process removing drug from the blood. A two-compartment model describes this type of serum concentration-time profile. The conduction system of the heart responds to the high concentrations of lidocaine present during the distribution phase, so lidocaine has a quick onset of action.
The generally accepted therapeutic range for lidocaine is 1.5-5 μg/mL. In the upper end of the therapeutic range (>3 μg/mL) some patients will experience minor side effects including drowsiness, dizziness, paresthesias, or euphoria. Lidocaine serum concentrations above the therapeutic range can cause muscle twitching, confusion, agitation, dysarthria, psychosis, seizures, or coma. Cardiovascular adverse effects such as atrioventricular block, hypotension, and circulatory collapse have been reported at lidocaine concentrations above 6 μg/mL, but are not strongly correlated with specific serum levels. Lidocaine-induced seizures are not as difficult to treat as theophylline-induced seizures and usually respond to traditional antiseizure medication therapy. Lidocaine metabolites (MEGX and GX, see Basic Clinical Pharmacokinetic Parameter section) probably contribute to the central nervous system side effects attributed to lidocaine therapy.6–8 Clinicians should understand that all patients with “toxic” lidocaine serum concentrations in the listed ranges will not exhibit signs or symptoms of lidocaine toxicity. Rather, lidocaine concentrations in the given ranges increase the likelihood that an adverse effect will occur.
For dose adjustment purposes, lidocaine serum concentrations are best measured at steady-state after the patient has received a consistent dosage regimen for 3-5 drug half-lives. Lidocaine half-life varies from 1 to 1.5 hours in normal adults to 5 hours or more in adult patients with liver failure. If lidocaine is given as a continuous intravenous infusion, it can take a considerable amount of time (3-5 half-lives or 7.5-25 hours) for patients to achieve effective concentrations, so an intravenous loading dose is commonly administered to patients (Figure 7-2). The ideal situation is to administer an intravenous loading dose that will achieve the desired concentration immediately, then start an intravenous continuous infusion that will maintain that concentration (Figure 7-2). In order to derive this perfect situation, the lidocaine volume of distribution for the central compartment (Vc in L) would have to be known to compute the loading dose (LD in mg): LD = Css • Vc, where Css is the desired lidocaine concentration in mg/L. The volume of distribution for the central compartment of the two-compartment model is used to compute the loading dose because lidocaine distributes rapidly to the myocardium and the heart is considered to reside in the central compartment of the model. However, this pharmacokinetic parameter is rarely, if ever, known for a patient, so a loading dose based on a population average central volume of distribution is used to calculate the amount of lidocaine needed. Since the patient’s own, unique central volume of distribution will most likely be greater (resulting in too low of a loading dose) or less (resulting in too large of a loading dose) than the population average volume of distribution used to compute the loading dose, the desired steady-state lidocaine concentration will not be achieved. Because of this, it will still take 3-5 half-lives for the patient to reach steady-state conditions while receiving a constant intravenous infusion rate (Figure 7-3).
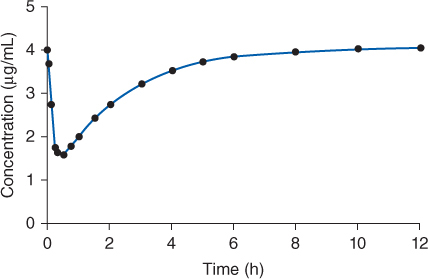
Figure 7-2 To maintain therapeutic lidocaine concentrations, an intravenous bolus (over 1-5 minutes) of lidocaine is followed by a continuous intravenous infusion of the drug. Even though the infusion is started right after the loading dose is given, serum concentrations due to the infusion cannot increase rapidly enough to counter the large decrease in concentrations during the distribution phase from the bolus dose. The dip in serum lidocaine concentrations below therapeutic amounts can allow previously treated arrhythmias to recur.
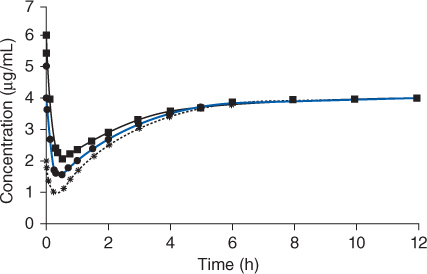
Figure 7-3 Because the central volume of distribution is not known at the time an intravenous loading dose of lidocaine is administered, average population parameters must be assumed and almost always result in initial lidocaine serum concentrations that are higher (dashed line with squares) or lower (dotted line with triangles) than those that were expected (solid line with circles). So, the main clinical goal of administering loading doses of lidocaine is to achieve therapeutic concentrations as soon as possible, not to attain steady-state concentrations immediately after the loading dose is given.
After a lidocaine loading dose is given, serum concentrations from this dose rapidly decline due to distribution from blood to tissues, and serum concentrations due to the infusion are not able to increase rapidly enough to avoid a temporary decline or dip in lidocaine concentrations (Figure 7-2). The decline may be severe enough that ventricular arrhythmias which were initially suppressed by lidocaine may recur due to subtherapeutic antiarrhythmic concentrations. Because of this dip in concentrations due to distribution of drug after the intravenous loading dose, an additional dose (50% of original loading dose) can be given 20-30 minutes after the original loading dose. Or, several additional doses (33%-50% of original loading dose) can be given every 5-10 minutes to a total maximum of 3 mg/kg (Figure 7-4).5 Thus, lidocaine intravenous loading doses do not usually achieve steady-state serum concentrations immediately, but, hopefully, they do result in therapeutic concentrations and response sooner than simply starting an intravenous infusion alone.
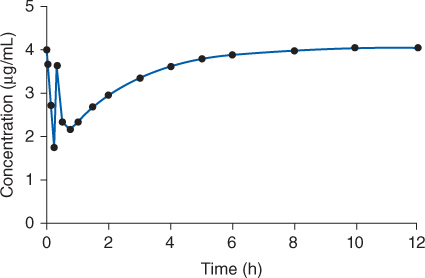
Figure 7-4 Since the dip in serum lidocaine concentrations below therapeutic amounts can allow previously treated arrhythmias to recur, a supplemental loading or “booster” dose is typically given 20-30 minutes after the initial loading dose. This prevents lidocaine serum concentrations from declining too far during the distribution phase of the intravenous bolus dose and before serum concentrations from the intravenous infusion have an opportunity to attain therapeutic concentrations.
CLINICAL MONITORING PARAMETERS
The electrocardiogram (ECG or EKG) should be monitored to determine the response to lidocaine in patients with ventricular tachycardia. The goal of therapy is suppression of ventricular arrhythmias and avoidance of adverse drug reactions. Lidocaine therapy is often discontinued after 6-24 hours of treatment so the need for long-term antiarrhythmic drug use can be reassessed, although longer infusions may be used in patients with persistent tachyarrhythmias. For long-term therapy, electrophysiologic studies using programmed stimulation to replicate the ventricular arrhythmia or 24-hour ECG monitoring using a Holter monitor can be performed in patients while receiving a variety of antiarrhythmic agents to determine effective antiarrhythmic drug therapy. Because lidocaine is only administered parenterally, it is rarely used for more than a few days unless oral antiarrhythmic agents are ineffective.
Because lidocaine is usually given for a short duration (<24 hours), it is often not necessary to obtain serum lidocaine concentrations in patients receiving appropriate doses who currently have no ventricular arrhythmia or adverse drug effects. However, lidocaine serum concentrations should be obtained in patients who have a recurrence of ventricular tachyarrhythmias, are experiencing possible lidocaine side effects, or are receiving lidocaine doses not consistent with disease states and conditions known to alter lidocaine pharmacokinetics (see Effects of Disease States and Conditions on Lidocaine Pharmacokinetics and Dosing section). Serum concentration monitoring can aid in the decision to increase or decrease the lidocaine dose. For instance, if the ventricular arrhythmia reappears and the lidocaine serum concentration is <5 μg/mL, increasing the lidocaine dose is a therapeutic option. However, if the lidocaine serum concentration is over 5 μg/mL, it is unlikely that a dosage increase will be effective in suppressing the arrhythmia and there is an increased likelihood that drug side effects may occur. Similarly, if a possible lidocaine adverse drug reaction is noted in a patient and the lidocaine serum concentration is <3-5 μg/mL, it is possible that the observed problem may not be due to lidocaine treatment and other sources can be investigated. Patients receiving lidocaine infusions for longer than 24 hours are prone to unexpected accumulation of lidocaine concentrations in the serum and should be closely monitored for lidocaine side effects.9–12 While receiving lidocaine, patients should be monitored for the following adverse drug effects: drowsiness, dizziness, paresthesias, euphoria, muscle twitching, confusion, agitation, dysarthria, psychosis, seizures, coma, atrioventricular block, or hypotension.
BASIC CLINICAL PHARMACOKINETIC PARAMETERS
Lidocaine is almost completely eliminated by hepatic metabolism (>95%).4,13 Hepatic metabolism is mainly via the CYP3A enzyme system. Monoethylglycinexylidide (MEGX) is the primary metabolite resulting from lidocaine metabolism.6–8 While a portion of MEGX is eliminated renally, most of the metabolite is further converted hepatically to glycinexylidide (GX) and other, inactive metabolites. GX is primarily eliminated by the kidney. MEGX and GX have some antiarrhythmic activity (MEGX ~80% and GX ~10%, relative to lidocaine), but have also been implicated as the cause of some adverse effects attributed to lidocaine therapy.6–8 Because both metabolites are eliminated by the kidney, patients with renal failure should be monitored for adverse effects due to metabolite accumulation even though lidocaine serum concentrations are within the therapeutic range. The hepatic extraction ratio of lidocaine is about 70%, so lidocaine is typically classified as a high extraction ratio drug. Because of this, it is expected that liver blood flow will be the predominate factor influencing the clearance of lidocaine (Cl ≈ LBF, where Cl is lidocaine clearance and LBF is liver blood flow, both in L/min), and many disease states and conditions that alter lidocaine clearance do so via changes in liver blood flow. However, because a hepatic extraction ratio greater than 70% is the definition of a high extraction ratio agent and the extraction ratio for lidocaine is on the margin of this range, it is very possible that changes in lidocaine intrinsic clearance or plasma protein binding will change lidocaine clearance.
Lidocaine is usually given intravenously but may also be given intramuscularly.14 After intramuscular injection, absorption is rapid and complete with maximum concentrations occurring about 1 hour after administration and 100% bioavailability as long as the patient’s peripheral circulation is not compromised due to hypotension or shock. Intramuscular administration of medications can increase creatine kinase (CK) concentrations due to minor skeletal muscle trauma inflicted by the injection, and this enzyme is monitored in patients who may have had a myocardial infarction. Thus, the creatine kinase isozyme that is relatively specific to the heart (CK-MB) needs to be measured in myocardial infarction patients who have received intramuscular injections. Oral absorption of lidocaine is nearly 100%.4 However, lidocaine is extensively metabolized by the CYP3A enzymes contained in the intestinal wall and liver resulting in a large first-pass effect and low, variable oral bioavailability (F ≈ 30%). Because roughly 70% of an oral dose is converted to metabolites, MEGX and GX concentrations are high after oral administration of lidocaine resulting in a high incidence of adverse effects.
Lidocaine can also be administered intraosseously or endotracheally. Although the optimal dose of lidocaine administered via an endotracheal tube is not known, it is typically 2-2.5 times the recommended intravenous dose.3 Lidocaine is given through an endotracheal tube by first diluting the drug with an additional 5-10 mL of sterile water or normal saline, then injecting the entire amount into the tube.
Plasma protein binding in normal individuals is about 70%.15–17 Of this value, approximately 30% is due to drug binding to albumin while 70% is due to lidocaine bound to α1-acid glycoprotein (AGP).9,11,12 AGP is classified as an acute phase reactant protein that is present in lower amounts in all individuals but is secreted in large amounts in response to certain stresses and disease states such as trauma, heart failure, and myocardial infarction. In patients with these disease states, lidocaine binding to AGP can be even larger resulting in an unbound fraction as low as 10%-15%. AGP concentrations continuously increase during the first 12-72 hours after a myocardial infarction, and, as a result, the lidocaine-unbound fraction decreases on average from about 30% to 20% during this time period. The continuous increase in protein binding due to AGP secretion causes a continuous decrease in lidocaine clearance in patients with myocardial infarction, and lidocaine concentrations can accumulate to unexpectedly high levels in patients receiving the drug for longer than 24 hours. Patients without myocardial infarction also experience accumulation of lidocaine concentrations during long-term (>24 hours) infusions due to competition for hepatic metabolism between parent drug and metabolites.10,18 Thus, monitoring for adverse reactions in patients receiving long-term lidocaine infusions is important, and lidocaine serum concentrations can be useful adjuncts to avoid lidocaine toxicity.
The recommended dose of lidocaine is based on the concurrent disease states and conditions present in the patient that can influence lidocaine concentrations. Lidocaine pharmacokinetic parameters used to compute doses are given in the following section for specific patient profiles.
EFFECTS OF DISEASE STATES AND CONDITIONS ON LIDOCAINE PHARMACOKINETICS AND DOSING
Normal adults without the disease states and conditions given later in this section with normal liver function have an average lidocaine half-life of 1.5 hours (range: 1-2 hours), a central volume of distribution of 0.5 L/kg (Vc = 0.4-0.6 L/kg), and the volume of distribution for the entire body of 1.5 L/kg (Varea = 1-2 L/kg; Table 7-1).4,10,19 Disease states and conditions that change lidocaine pharmacokinetics and dosage requirements may alter clearance, the central volume of distribution, and the volume of distribution for the entire body. The volume of distribution for the central compartment of the two-compartment model is used to compute the loading dose because lidocaine distributes rapidly to the myocardium and the heart is considered to reside in the central compartment of the model. The elimination rate constant (k = 0.693/t1/2, where t1/2 is the half-life) and clearance (Cl = kVarea) can be computed from the aforementioned pharmacokinetic parameters.
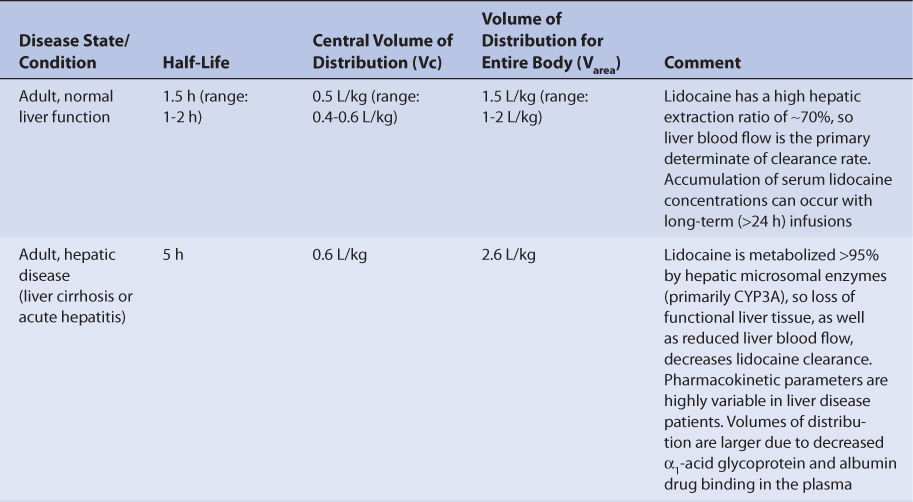
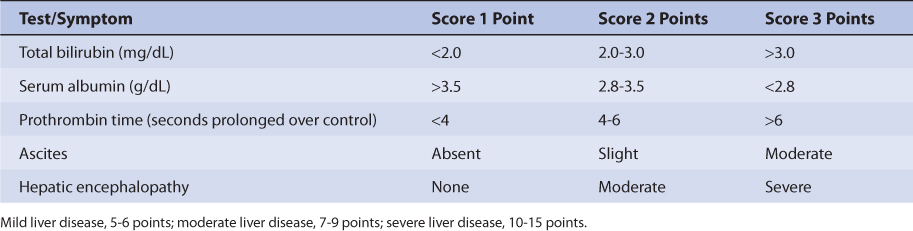
Patients with liver cirrhosis or acute hepatitis have reduced lidocaine clearance which results in a prolonged average lidocaine half-life of 5 hours.13,20–23 The mechanism for depressed clearance in liver disease patients is destruction of liver parenchyma, where hepatic drug-metabolizing enzymes are present, and reduction of liver blood flow. The central volume of distribution and volume of distribution for the entire body are larger in patients with liver disease because albumin and α1-acid glycoprotein (AGP) concentrations are lower in these patients and result in reduced lidocaine plasma protein binding (average Vc = 0.6 L/kg, average Varea = 2.6 L/kg). However, the effect that liver disease has on lidocaine pharmacokinetics is highly variable and difficult to accurately predict, especially in patients with acute hepatitis. It is possible for a patient with liver disease to have relatively normal or grossly abnormal lidocaine clearance, volumes of distribution, and half-life. An index of liver dysfunction can be gained by applying the Child-Pugh clinical classification system to the patient (Table 7-2).24 Child-Pugh scores are completely discussed in Chapter 3 (Drug Dosing in Special Populations: Renal and Hepatic Disease, Dialysis, Heart Failure, Obesity, and Drug Interactions), but will be briefly discussed here. The Child-Pugh score consists of five laboratory tests or clinical symptoms: serum albumin, total bilirubin, prothrombin time, ascites, and hepatic encephalopathy. Each of these areas is given a score of 1 (normal) to 3 (severely abnormal; Table 7-2), and the scores for the five areas are summed. The Child-Pugh score for a patient with normal liver function is 5 while the score for a patient with grossly abnormal serum albumin, total bilirubin, and prothrombin time values in addition to severe ascites and hepatic encephalopathy is 15. A Child-Pugh score greater than 8 is the ground for a decrease in the initial daily drug dose for lidocaine (t1/2 = 5 hours). As in any patient with or without liver dysfunction, initial doses are meant as starting points for dosage titration based on patient response and avoidance of adverse effects. Lidocaine serum concentrations and the presence of adverse drug effects should be monitored frequently in patients with liver cirrhosis.
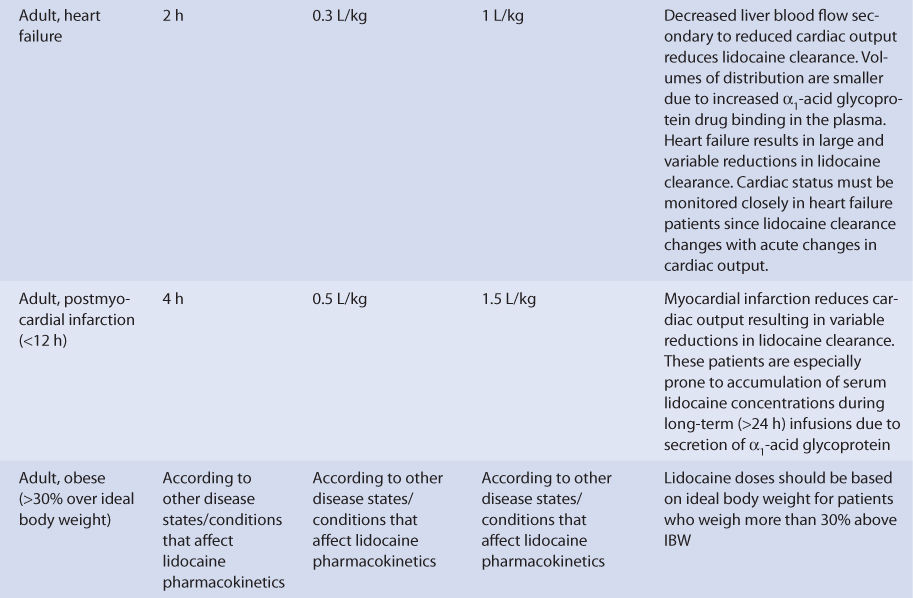
Heart failure causes reduced lidocaine clearance because of decreased hepatic blood flow secondary to compromised cardiac output (Table 7-3).6,13,22,25,26 Patients with cardiogenic shock experience extreme declines in lidocaine clearance due to severe decreases in cardiac output and liver blood flow. Central volume of distribution (Vc = 0.3 L/kg) and volume of distribution for the entire body (Varea = 1 L/kg) are decreased because heart failure patients have elevated AGP serum concentrations which lead to increased lidocaine plasma protein binding and decreased lidocaine unbound fraction. Patients with heart failure have an average lidocaine half-life equal to 2 hours (range: 1-24 hours). Half-life (t1/2) does not change as much as expected from the change in clearance (Cl) because the volume of distribution simultaneously decreases [t1/2 = (0.693 • ↓Varea)/↓Cl]. Obviously, the effect that heart failure has on lidocaine pharmacokinetics is highly variable and difficult to accurately predict. It is possible for a patient with heart failure to have relatively normal or grossly abnormal lidocaine clearance and half-life. For heart failure patients, initial doses are meant as starting points for dosage titration based on patient response and avoidance of adverse effects. Lidocaine serum concentrations and the presence of adverse drug effects should be monitored frequently in patients with heart failure.

Patients with myocardial infarction may develop serious ventricular arrhythmias that require therapy with lidocaine. After a myocardial infarction, serum AGP concentrations increase up to 50% over a 12-72-hour time period.9,11,12 As AGP serum concentrations increase, plasma protein binding of lidocaine decreases and the unbound fraction of lidocaine decreases from about 30% to about 20%. Although lidocaine is considered a high hepatic extraction ratio drug with liver blood flow having the major influence on lidocaine clearance, a decline in the unbound fraction of lidocaine in the plasma decreases lidocaine clearance. The reduction in lidocaine clearance is continuous as long as AGP concentrations continue to rise. A result of this phenomenon is lidocaine serum concentrations do not reach steady-state during long-term (>24 h) intravenous infusions of lidocaine in myocardial infarction patients and results of pharmacokinetic studies in this patient population differ according to when the investigation took place in relation to the myocardial damage. When studied within 12 hours of myocardial infarction, patients had decreased lidocaine clearance due to decreased cardiac output and liver blood flow, relatively normal volumes of distribution (Vc = 0.5 L/kg, Varea = 1.5 L/kg), and a prolonged half-life of 4 hours.26–28 When similar myocardial infarction patients are studied after longer lidocaine infusions, the central volume of distribution and volume of distribution representing the entire body are smaller because AGP serum concentrations have had an opportunity to increase and change lidocaine plasma protein binding.9,11,12
Although the volume of distribution representing the entire body (Varea) correlates most closely with total body weight, obese patients (>30% above ideal body weight or IBW) should have central volume of distribution and clearance estimates based on ideal body weight.19 Lidocaine pharmacokinetic parameter estimates should be based on the concurrent disease states and conditions present in the patient. If weight-based dosage recommendations are to be used, ideal body weight should be used to compute maintenance infusions (mg/kg/min) and loading doses (mg/kg) for obese individuals.
Patient age has an effect on lidocaine volumes of distribution and half-life.25 For elderly patients over the age of 65, studies indicate that lidocaine clearance is unchanged, the volumes of distribution are slightly larger, and half-life is longer (average half-life = 2.3 hours, range: 1.7-4.5 hours) compared to younger subjects. A confounding factor found in lidocaine pharmacokinetic studies conducted in older adults is the possible accidental inclusion of subjects that have subclinical or mild cases of the disease states associated with reduced lidocaine clearance (heart failure, liver disease, etc). Additionally, most patients with serious ventricular arrhythmias studied in all of the previously mentioned studies are older and those results include any influence that age may have. Thus, in most cases elderly patients are treated with lidocaine according to the other disease states or conditions present that influence lidocaine pharmacokinetics.
Lidocaine serum concentrations accumulate in patients receiving long-term (>24 h) infusions even if the patient did not have a myocardial infarction.10,18 Accumulation of lidocaine in these patients is due to competition for hepatic metabolism between parent drug and metabolites. Because MEGX and GX metabolites are eliminated to some extent by the kidney, patients with renal failure should be monitored for lidocaine adverse effects due to metabolite accumulation even though lidocaine serum concentrations are within the therapeutic range. Lidocaine is not appreciably removed by hemodialysis. Because lidocaine has a sieving coefficient of 0.14, continuous hemofiltration does not remove a significant amount of drug.29,30
DRUG INTERACTIONS
Lidocaine has serious drug interactions with β-adrenergic receptor blockers and cimetidine that decrease lidocaine clearance by 30% or more.31 Propranolol, metoprolol, and nadolol have been reported to reduce lidocaine clearance due to the decrease in cardiac output caused by β-blocker agents. Decreased cardiac output results in reduced liver blood flow which explains the decline in lidocaine clearance caused by these drugs. Cimetidine also decreases lidocaine clearance, but the mechanism of the interaction is different. Because cimetidine does not change liver blood flow, it is believed that cimetidine decreases lidocaine clearance by inhibiting hepatic microsomal enzymes.32,33
Lidocaine clearance may be accelerated by concomitant use of phenobarbital or phenytoin.31 Both of these agents are known to be hepatic drug-metabolizing enzyme inducers, and this is the probable mechanism of their drug interaction with lidocaine. It is important to remember that phenytoin has antiarrhythmic effects and is also classified as a type IB antiarrhythmic agent. Because of this, phenytoin and lidocaine may have additive pharmacologic effects that could result in a pharmacodynamic drug interaction.
INITIAL DOSAGE DETERMINATION METHODS
Several methods to initiate lidocaine therapy are available. The pharmacokinetic dosing method is the most flexible of the techniques. It allows individualized target serum concentrations to be chosen for a patient, and each pharmacokinetic parameter can be customized to reflect specific disease states and conditions present in the patient. Literature-based recommended dosing is a very commonly used method to prescribe initial doses of lidocaine. Doses are based on those that commonly produce steady-state concentrations in the lower end of the therapeutic range, although there is a wide variation in the actual concentrations for a specific patient.
Pharmacokinetic Dosing Method
The goal of initial dosing of lidocaine is to compute the best dose possible for the patient given their set of disease states and conditions that influence lidocaine pharmacokinetics and the arrhythmia being treated. In order to do this, pharmacokinetic parameters for the patient will be estimated using average parameters measured in other patients with similar disease state and condition profiles.
Half-Life and Elimination Rate Constant Estimate
Lidocaine is predominately metabolized by liver. Unfortunately, there is no good way to estimate the elimination characteristics of liver-metabolized drugs using an endogenous marker of liver function in the same manner that serum creatinine and estimated creatinine clearance are used to estimate the elimination of agents that are renally eliminated. Because of this, a patient is categorized according to the disease states and conditions that are known to change lidocaine half-life, and the half-life previously measured in these studies is used as an estimate of the current patient’s half-life (Table 7-1). For example, if a patient has suffered an uncomplicated myocardial infarction, lidocaine half-life would be assumed to equal 4 hours. Alternatively, for a patient with moderate heart failure (NYHA CHF class III), lidocaine half-life would be assumed to equal 2 hours, while a patient with severe liver disease (Child-Pugh score = 12) would be assigned an estimated half-life of 5 hours. To produce the most conservative lidocaine doses in patients with multiple concurrent disease states or conditions that affect lidocaine pharmacokinetics, the disease state or condition with the longest half-life should be used to compute doses. This approach will avoid accidental overdosage as much as currently possible. Once the correct half-life is identified for the patient, it can be converted into the lidocaine elimination rate constant (k) using the following equation: k = 0.693/t1/2.
Volume of Distribution Estimate
As with the half-life estimate, lidocaine volume of distribution values are chosen according to the disease states and conditions that are present (Table 7-1). The central volume of distribution (Vc) is used to compute loading doses because lidocaine has a rapid onset of action after administration, and the heart acts as if it is in the central compartment of the two-compartment model used to describe lidocaine pharmacokinetics. The central volume of distribution is assumed to equal 0.6 L/kg for liver disease patients, 0.3 L/kg for heart failure and cardiogenic shock patients, and 0.5 L/kg for all other patients. The volume of distribution for the entire body after distribution is complete (Varea) is used to help compute lidocaine clearance, and is assumed to equal 2.6 L/kg for liver disease patients, 1 L/kg for heart failure and cardiogenic shock patients, and 1.5 L/kg for all other patients. For obese patients (>30% above ideal body weight), ideal body weight is used to compute lidocaine volume of distribution. Thus, for a nonobese 80 kg patient without heart failure or liver disease, the estimated lidocaine central volume of distribution would be 40 L: Vc = 0.5 L/kg • 80 kg = 40 L. For a 150-kg obese patient with an ideal body weight of 60 kg and normal cardiac and liver function, the estimated lidocaine volume of distribution is 30 L: V = 0.5 L/kg • 60 kg = 30 L.
Selection of Appropriate Pharmacokinetic Model and Equations
When given by continuous intravenous infusion, lidocaine follows a two-compartment pharmacokinetic model (Figures 7-1, 7-2, 7-3). A simple pharmacokinetic equation that computes the lidocaine steady-state serum concentration (Css in μg/mL = mg/L) is widely used and allows dosage calculation for a continuous infusion: Css = k0/Cl or k0 = Css • Cl, where k0 is the dose of lidocaine in mg and Cl is lidocaine clearance in L/h. Clearance is computed using estimates of lidocaine elimination rate constant (k) and volume of distribution for the entire body after distribution is complete (Varea): Cl = kVarea. For example, if a patient has an estimated elimination rate constant equal to 0.173 h–1 and an estimated volume of distribution equal to 105 L, the estimated clearance would equal 18.2 L/h: Cl = 0.173 h–1 • 105 L = 18.2 L/h.
The equation used to calculate an intravenous loading dose (LD in mg) is based on a two-compartment model: LD = (Css • Vc), where Css is the desired lidocaine steady-state concentration in μg/mL which is equivalent to mg/L, and Vc is the lidocaine central volume of distribution. Intravenous lidocaine loading doses should be given as an intravenous bolus no faster than 25-50 mg/min.
Steady-State Concentration Selection
The general accepted therapeutic range for lidocaine is 1.5-5 μg/mL. However, lidocaine therapy must be individualized for each patient in order to achieve optimal responses and minimal side effects.

LK is a 50-year-old, 75-kg (height = 5 ft 10 in) male with ventricular tachycardia who requires therapy with intravenous lidocaine. He has normal liver and cardiac function. Suggest an initial intravenous lidocaine dosage regimen designed to achieve a steady-state lidocaine concentration equal to 3 μg/mL.
1. Estimate the half-life and elimination rate constant according to disease states and conditions present in the patient.
The expected lidocaine half-life (t1/2) is 1.5 hours. The elimination rate constant is computed using the following formula: k = 0.693/t1/2 = 0.693/1.5 h = 0.462 h−1.
2. Estimate volume of distribution and clearance.
The patient is not obese, so the estimated lidocaine central volume of distribution and the volume of distribution for the entire body (Varea) will be based on actual body weight: Vc = 0.5 L/kg • 75 kg = 38 L, Varea = 1.5 L/kg • 75 kg = 113 L. Estimated lidocaine clearance is computed by taking the product of Varea and the elimination rate constant: Cl = kVarea = 0.462 h−1 • 113 L = 52.2 L/h.
3. Compute dosage regimen.
Therapy will be started by administering an intravenous loading dose of lidocaine to the patient (note: μg/mL = mg/L and this concentration unit was substituted for Css in the calculations so that unnecessary unit conversion was not required): LD = Css • Vc = 3 mg/L • 38 L = 114 mg, rounded to 100 mg intravenously over 2-4 minutes. An additional dose equal to 50% of the loading dose can be given if arrhythmias recur 20-30 minutes after the initial loading dose.
A lidocaine continuous intravenous infusion will be started immediately after the loading dose has been administered. (Note: μg/mL = mg/L and this concentration unit was substituted for Css in the calculations so that unnecessary unit conversion was not required.) The dosage equation for intravenous lidocaine is: k0 = Css • Cl = (3 mg/L • 52.2 L/h)/(60 min/h) = 2.6 mg/min, rounded to 2.5 mg/min.
A steady-state lidocaine serum concentration could be measured after steady-state is attained in 3-5 half-lives. Since the patient is expected to have a half-life equal to 1.5 hours, the lidocaine steady-state concentration could be obtained any time after the first 8 hours of dosing (5 half-lives = 5 • 1.5 h = 7.5 h). Lidocaine serum concentrations should also be measured if the patient experiences a return of their ventricular arrhythmia, or if the patient develops potential signs or symptoms of lidocaine toxicity.
EXAMPLE 2 
OI is a 60-year-old, 85-kg (height = 6 ft 1 in) male with ventricular tachycardia who requires therapy with intravenous lidocaine. He has liver cirrhosis (Child-Pugh score = 11). Suggest an initial intravenous lidocaine dosage regimen designed to achieve a steady-state lidocaine concentration equal to 4 μg/mL.
1. Estimate the half-life and elimination rate constant according to disease states and conditions present in the patient.
The expected lidocaine half-life (t1/2) is 5 hours. The elimination rate constant is computed using the following formula: k = 0.693/t1/2 = 0.693/5 h = 0.139 h–1.
2. Estimate volume of distribution and clearance.
The patient is not obese, so the estimated lidocaine central volume of distribution and the volume of distribution for the entire body (Varea) will be based on actual body weight: Vc = 0.6 L/kg • 85 kg = 51 L, Varea = 2.6 L/kg • 85 kg = 221 L. Estimated lidocaine clearance is computed by taking the product of Varea and the elimination rate constant: Cl = kVarea = 0.139 h−1 • 221 L = 31 L/h.
Therapy will be started by administering an intravenous loading dose of lidocaine to the patient (note: μg/mL = mg/L and this concentration unit was substituted for Css in the calculations so that unnecessary unit conversion was not required): LD = Css • Vc = 4 mg/L • 51 L = 204 mg, rounded to 200 mg intravenously over 4-8 minutes. An additional dose equal to 50% of the loading dose can be given if arrhythmias recur 20-30 minutes after the initial loading dose.
A lidocaine continuous intravenous infusion will be started immediately after the loading dose has been administered. (Note: μg/mL = mg/L and this concentration unit was substituted for Css in the calculations so that unnecessary unit conversion was not required.) The dosage equation for intravenous lidocaine is: k0 = Css • Cl = (4 mg/L • 31 L/h)/(60 min/h) = 2.1 mg/min, rounded to 2 mg/min.
A steady-state lidocaine serum concentration could be measured after steady-state is attained in 3-5 half-lives. Since the patient is expected to have a half-life equal to 5 hours, the lidocaine steady-state concentration could be obtained any time after the first day of dosing (5 half-lives = 5 • 5 h = 25 h). Lidocaine serum concentrations should also be measured if the patient experiences a return of their ventricular arrhythmia, or if the patient develops potential signs or symptoms of lidocaine toxicity.
EXAMPLE 3 
MN is a 64-year-old, 78-kg (height = 5 ft 9 in) male with ventricular tachycardia who requires therapy with intravenous lidocaine. He has moderate heart failure (NYHA CHF class III). Suggest an initial intravenous lidocaine dosage regimen designed to achieve a steady-state lidocaine concentration equal to 3 μg/mL.
1. Estimate the half-life and elimination rate constant according to disease states and conditions present in the patient.
The expected lidocaine half-life (t1/2) is 2 hours. The elimination rate constant is computed using the following formula: k = 0.693/t1/2 = 0.693/2 h = 0.347 h–1.
2. Estimate volume of distribution and clearance.
The patient is not obese, so the estimated lidocaine central volume of distribution and the volume of distribution for the entire body (Varea) will be based on actual body weight: Vc = 0.3 L/kg • 78 kg = 23 L, Varea = 1 L/kg • 78 kg = 78 L. Estimated lidocaine clearance is computed by taking the product of Varea and the elimination rate constant: Cl = kVarea = 0.347 h–1 • 78 L = 27 L/h.
3. Compute dosage regimen.
Therapy will be started by administering an intravenous loading dose of lidocaine to the patient (note: μg/mL = mg/L and this concentration unit was substituted for Css in the calculations so that unnecessary unit conversion was not required): LD = Css • Vc = 3 mg/L • 23 L = 69 mg, rounded to 75 mg intravenously over 2-3 minutes. An additional dose equal to 50% of the loading dose can be given if arrhythmias recur 20-30 minutes after the initial loading dose.
A lidocaine continuous intravenous infusion will be started immediately after the loading dose has been administered. (Note: μg/mL = mg/L and this concentration unit was substituted for Css in the calculations so that unnecessary unit conversion was not required.) The dosage equation for intravenous lidocaine is: k0 = Css • Cl = (3 mg/L • 27 L/h)/(60 min/h) = 1.4 mg/min, rounded to 1.5 mg/min.
A steady-state lidocaine serum concentration could be measured after steady-state is attained in 3-5 half-lives. Since the patient is expected to have a half-life equal to 1.5 hours, the lidocaine steady-state concentration could be obtained any time after the first 10-12 hours of dosing (5 half-lives = 5 • 2 h = 10 h). Lidocaine serum concentrations should also be measured if the patient experiences a return of their ventricular arrhythmia, or if the patient develops potential signs or symptoms of lidocaine toxicity.
Literature-Based Recommended Dosing
Because of the large amount of variability in lidocaine pharmacokinetics, even when concurrent disease states and conditions are identified, many clinicians believe that the use of standard lidocaine doses for various situations is warranted.34 The original computation of these doses were based on the pharmacokinetic dosing method described in the previous section, and subsequently modified based on clinical experience. In general, the lidocaine steady-state serum concentration expected from the lower end of the dosage range was 1.5-3 μg/mL and 3-5 μg/mL for the upper end of the dosage range. Suggested intravenous lidocaine continuous infusion maintenance doses are 1-2 mg/min for patients with liver disease or heart failure and 3-4 mg/min for all other patients. When more than one disease state or condition is present in a patient, choosing the lowest infusion rate will result in the safest, most conservative dosage recommendation. With regard to loading doses, lidocaine is given intravenously at the dose of 1-1.5 mg/kg (not to exceed 25-50 mg/min) for all patients except those with heart failure. The suggested lidocaine intravenous loading dose for heart failure patients is 0.5-0.75 mg/kg (not to exceed 25-50 mg/min), although some clinicians advocate the administration of full loading doses of lidocaine in heart failure patients. Ideal body weight is used to compute loading doses for obese patients (>30% over ideal body weight).
Pediatric doses are similar to that given to adults when adjusted for differences in body weight. Intravenous loading doses are 1 mg/kg with up to two additional doses, if needed (total dose not to exceed 3-5 mg/kg for first hour). Continuous intravenous infusions doses are 20-50 μg/kg/min. For patients with shock, heart failure, or liver disease patients, initial doses should not exceed 20 μg/kg/min.35
To illustrate the similarities and differences between this method of dosage calculation and the Pharmacokinetic Dosing method, the same examples used in the previous section will be used.
EXAMPLE 1 
LK is a 50-year-old, 75-kg (height = 5 ft 10 in) male with ventricular tachycardia who requires therapy with intravenous lidocaine. He has normal liver and cardiac function. Suggest an initial intravenous lidocaine dosage regimen designed to achieve a steady-state lidocaine concentration equal to 3 μg/mL.
1. Choose a lidocaine dose based on disease states and conditions present in the patient.
A lidocaine loading dose of 1-1.5 mg/kg and maintenance infusion of 3-4 mg/min is suggested for a patient with heart failure.
2. Compute dosage regimen.
Because the desired concentration is in the lower end of the therapeutic range, a dose in the lower end of the suggested ranges will be used. A lidocaine loading dose of 1 mg/kg will be administered: LD = 1 mg/kg • 75 kg = 75 mg over 1-5-3 minutes. A lidocaine maintenance infusion equal to 3 mg/min would be administered after the loading dose was given. An additional dose equal to 50% of the loading dose can be given if arrhythmias recur 20-30 minutes after the initial loading dose.
A steady-state lidocaine serum concentration could be measured after steady-state is attained in 3-5 half-lives. Since the patient is expected to have a half-life equal to 1.5 hours, the lidocaine steady-state concentration could be obtained any time after the first 8 hours of dosing (5 half-lives = 5 • 1.5 h = 7.5 h). Lidocaine serum concentrations should also be measured if the patient experiences a return of their ventricular arrhythmia, or if the patient develops potential signs or symptoms of lidocaine toxicity.

OI is a 60-year-old, 85 kg (height = 6 ft 1 in) male with ventricular tachycardia who requires therapy with intravenous lidocaine. He has liver cirrhosis (Child-Pugh score = 11). Suggest an initial intravenous lidocaine dosage regimen designed to achieve a steady-state lidocaine concentration equal to 4 μg/mL.
1. Choose the lidocaine dose based on disease states and conditions present in the patient.
A lidocaine loading dose of 0.5-0.75 mg/kg and maintenance infusion of 1-2 mg/min is suggested for a patient with liver disease.
2. Compute dosage regimen.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


