120e | Carcinoma of Unknown Primary |
Carcinoma of unknown primary (CUP) is a biopsy-proven malignancy for which the anatomic site of origin remains unidentified after an intensive search. CUP is one of the 10 most frequently diagnosed cancers worldwide, accounting for 3–5% of all cancers. Most investigators limit CUP to epithelial cancers and do not include lymphomas, metastatic melanomas, and metastatic sarcomas because these cancers have specific histology- and stage-based treatments that guide management.
The emergence of sophisticated imaging, robust immunohistochemistry (IHC), and genomic and proteomic tools has challenged the “unknown” designation. Additionally, effective targeted therapies in several cancers have moved the paradigm from empiricism to considering a personalized approach to CUP management. The reasons cancers present as CUP remain unclear. One hypothesis is that the primary tumor either regresses after seeding the metastasis or remains so small that it is not detected. It is possible that CUP falls on the continuum of cancer presentation where the primary has been contained or eliminated by the natural body defenses. Alternatively, CUP may represent a specific malignant event that results in an increase in metastatic spread or survival relative to the primary. Whether the CUP metastases truly define a clone that is genetically and phenotypically unique to this diagnosis remains to be determined.
CUP BIOLOGY
Studies looking for unique signature abnormalities in CUP tumors have not been positive. Abnormalities in chromosomes 1 and 12 and other complex cytogenetic abnormalities have been reported. Aneuploidy has been described in 70% of CUP patients with metastatic adenocarcinoma or undifferentiated carcinoma. The overexpression of various genes, including Ras, bcl-2 (40%), her-2 (11%), and p53 (26–53%), has been studied in CUP samples, but they have no effect on response to therapy or survival. The extent of angiogenesis in CUP relative to that in metastases from known primaries has also been evaluated, but no consistent findings have emerged. Using the Sequenom (SQM) Massarray platform, a study in consecutive CUP patients showed that the overall mutational rate was surprisingly low (18%). No “new” low-frequency mutations were found using a panel of mutations involving the P13K/AKT pathway, MEK pathway, receptors, and downstream effectors. Nevertheless, it is possible that newer “deep sequencing” techniques in select patients may yield consistent abnormalities.
CLINICAL EVALUATION
Initial CUP evaluation has two goals: search for the primary tumor based on pathologic evaluation of the metastases and determine the extent of disease. Obtaining a thorough medical history from CUP patients is essential, including paying particular attention to previous surgeries, removed lesions, and family medical history to assess potential hereditary cancers. Adequate physical examination, including a digital rectal examination in men and breast and pelvic examinations in women, should be performed based on clinical presentation.
Role of Serum Tumor Markers and Cytogenetics Most tumor markers, including CEA, CA-125, CA 19-9, and CA 15-3, when elevated, are nonspecific and not helpful in determining the primary tumor site. Men who present with adenocarcinoma and osteoblastic metastasis should undergo a prostate-specific antigen (PSA) test. In patients with undifferentiated or poorly differentiated carcinoma (especially with a midline tumor), elevated β-human chorionic gonadotropin (β-hCG) and α fetoprotein (AFP) levels suggest the possibility of an extragonadal germ cell (testicular) tumor. With the availability of IHC, cytogenetic studies are rarely needed.
Role of Imaging Studies In the absence of contraindications, a baseline IV contrast computed tomography (CT) scan of the chest, abdomen, and pelvis is the standard of care. This helps to search for the primary tumor, evaluate the extent of disease, and select the most accessible biopsy site. Older studies suggested that the primary tumor site is detected in 20–35% of patients who undergo a CT scan of the abdomen and pelvis, although by current definition, these patients do not have CUP. These studies also suggest a latent primary tumor prevalence of 20%; with more sophisticated imaging, this has decreased to ≤5% today.
Mammography should be performed in all women who present with metastatic adenocarcinoma, especially in those with adenocarcinoma and isolated axillary lymphadenopathy. Magnetic resonance imaging (MRI) of the breast is a follow-up modality in patients with axillary adenopathy and suspected occult primary breast carcinoma following a negative mammography and ultrasound. The results of these imaging modalities can influence surgical management; a negative breast MRI result predicts a low tumor yield at mastectomy.
A conventional workup for a squamous cell carcinoma and cervical CUP (neck lymphadenopathy with no known primary tumor) includes a CT scan or MRI and invasive studies, including indirect and direct laryngoscopy, bronchoscopy, and upper endoscopy. Ipsilateral (or bilateral) staging tonsillectomy has been recommended for these patients. 18-Fluorodeoxyglucose positron emission tomography (18-FDG-PET) scans are useful in this patient population and may help guide the biopsy; determine the extent of disease; facilitate the appropriate treatment, including planning radiation fields; and help with disease surveillance. A smaller radiation field encompassing the primary (when found) and metastatic adenopathy decreases the risk of chronic xerostomia. Several studies have evaluated the utility of PET in patients with squamous cervical CUP, and head and neck primary tumors were identified in ~21–30%.
The diagnostic contribution of PET to the evaluation of other CUP (outside of the neck adenopathy indication) remains controversial and is not routinely recommended. PET-CT can be helpful for patients who are candidates for surgical intervention for solitary metastatic disease because the presence of disease outside the primary site may affect surgical planning.
Invasive studies, including upper endoscopy, colonoscopy, and bronchoscopy, should be limited to symptomatic patients or those with laboratory, imaging, or pathologic abnormalities that suggest that these techniques will result in a high yield in finding a primary cancer.
Role of Pathologic Studies A detailed pathologic examination of the most accessible biopsied tissue specimen is mandatory in CUP patients. Pathologic evaluation typically consists of hematoxylin and eosin stains and immunohistochemical tests.
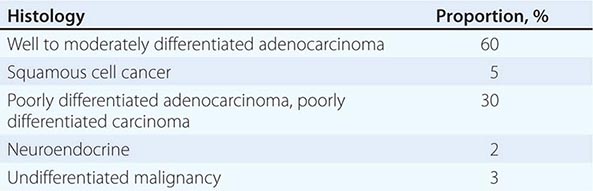
LIGHT MICROSCOPY EVALUATION Adequate tissue obtained preferably by excisional biopsy or core-needle biopsy (instead of only a fine-needle aspiration) is stained with hematoxylin and eosin and subjected to light microscopic examination. On light microscopy, 60–65% of CUP is adenocarcinoma, and 5% is squamous cell carcinoma. The remaining 30–35% is poorly differentiated adenocarcinoma, poorly differentiated carcinoma, or poorly differentiated neoplasm. A small percentage of lesions are diagnosed as neuroendocrine cancers (2%), mixed tumors (adenosquamous or sarcomatoid carcinomas), or undifferentiated neoplasms (Table 120e-1).
MAJOR HISTOLOGIES IN CARCINOMA OF UNKNOWN PRIMARY |
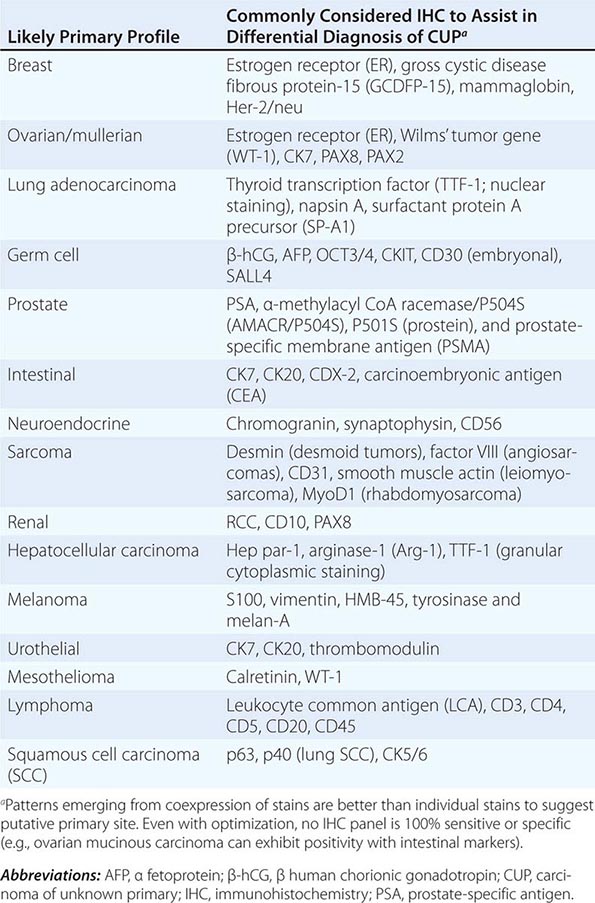
ROLE OF IMMUNOHISTOCHEMICAL ANALYSIS Immunohistochemical stains are peroxidase-labeled antibodies against specific tumor antigens that are used to define tumor lineage. The number of available immunohistochemical stains is ever-increasing. However, in CUP cases, more is not necessarily better, and immunohistochemical stains should be used in conjunction with the patient’s clinical presentation and imaging studies to select the best therapy. Communication between the clinician and pathologist is essential. No stain is 100% specific, and overinterpretation should be avoided. PSA and thyroglobulin tissue markers, which are positive in prostate and thyroid cancer, respectively, are the most specific of the current marker panel. However, these cancers rarely present as CUP, so the yield of these tests may be low. Fig. 120e-1 delineates a simple algorithm for immunohistochemical staining in CUP cases. Table 120e-2 lists additional tests that may be useful to further define the tumor lineage. A more comprehensive algorithm may improve the diagnostic accuracy but can make the process complex. With the use of immunohistochemical markers, electron microscopic analysis, which is time-consuming and expensive, is rarely needed.
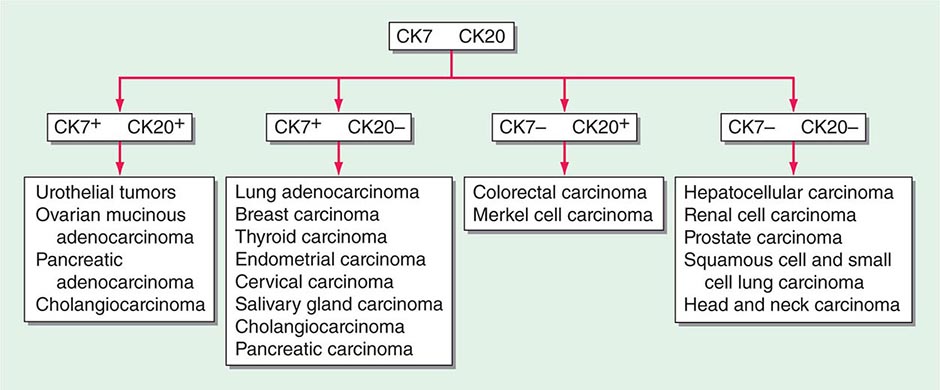
FIGURE 120e-1 Approach to cytokeratin (CK7 and CK20) markers used in adenocarcinoma of unknown primary.
SELECT IMMUNOHISTOCHEMICAL STAINS USEFUL IN THE DIAGNOSIS OF CARCINOMA OF UNKNOWN PRIMARY (CUP) |
There are >20 subtypes of cytokeratin (CK) intermediate filaments with different molecular weights and differential expression in various cell types and cancers. Monoclonal antibodies to specific CK subtypes have been used to help classify tumors according to their site of origin; commonly used CK stains in adenocarcinoma CUP are CK7 and CK20. CK7 is found in tumors of the lung, ovary, endometrium, breast, and upper gastrointestinal tract including pancreaticobiliary cancers, whereas CK20 is normally expressed in the gastrointestinal epithelium, urothelium, and Merkel cells. The nuclear CDX-2 transcription factor, which is the product of a homeobox gene necessary for intestinal organogenesis, is often used to aid in the diagnosis of gastrointestinal adenocarcinomas.
Thyroid transcription factor 1 (TTF-1) nuclear staining is typically positive in lung and thyroid cancers. Approximately 68% of adenocarcinomas and 25% of squamous cell lung cancers stain positive for TTF-1, which helps differentiate a lung primary tumor from metastatic adenocarcinoma in a pleural effusion, the mediastinum, or the lung parenchyma.
Gross cystic disease fibrous protein-15, a 15-kDa monomer protein, is a marker of apocrine differentiation that is detected in 62–72% of breast carcinomas. UROIII, high-molecular-weight cytokeratin, thrombomodulin, and CK20 are the markers used to diagnose lesions of urothelial origin.
IHC performs the best when used in groups that give rise to patterns that are strongly indicative of certain profiles. For example, the TTF-1/CK7+ and CK20+/CDX-2+/CK7– phenotypes have been reported as very suggestive of lung and lower gastrointestinal cancer profiles, respectively, although these patterns have not been validated prospectively in the absence of a primary cancer. IHC is not without its limitations; several factors affect tissue antigenicity (antigen retrieval, specimen processing, and fixation), interpretation of stains in tumor (nuclear, cytoplasmic, membrane) versus normal tissue, inter- and intraobserver variability, and tissue heterogeneity and inadequacy (given small biopsy sizes). Communication with the pathologist is critical to determine if additional tissue will be beneficial in the pathologic evaluation.
ROLE OF TISSUE OF ORIGIN MOLECULAR PROFILING In the absence of a known primary, developing therapeutic strategies for CUP is challenging. The current diagnostic yield with imaging and immunochemistry is ~20–30% for CUP patients. The use of gene expression studies holds the promise of substantially increasing this yield. Gene expression profiles are most commonly generated using quantitative reverse transcriptase polymerase chain reaction (RT-PCR) or DNA microarray.
Neural network programs have been used to develop predictive algorithms from the gene expression profiles. Typically, a training set of gene profiles from known cancers (preferably from metastatic sites) is used to train the software. The program can then be used to predict the putative origin of a test tumor and presumably of true CUP. Comprehensive gene expression databases that have become available for common malignancies may also be useful in CUP. These approaches have been effective in testing against known primary cancers and their metastases.
mRNA- or microRNA-based tissue of origin molecular profiling assays have been studied in prospective and retrospective CUP trials. Most of the CUP studies have evaluated assay performance, although the challenge with validating the accuracy of an assay for CUP is that, by definition, the primary cancer diagnosis cannot be verified. Thus, current estimates of tissue of origin test accuracy have relied on indirect metrics including comparison with IHC, clinical presentation, and appearance of latent primaries. Using these measures, the assays suggest a plausible primary in ~70% of patients studied. The only outcomes-based study is a single-arm study reporting a median survival of 12.5 months for patients who received assay-directed site-specific therapy. Firm conclusions of therapeutic impact cannot be drawn from this study given the nonrandomized design, statistical biases, confounding variables including use of subsequent lines of (empiric) therapy, and the heterogeneity of the CUP cancers. Additional studies are needed to better understand the clinical influence of tissue of origin profiling tools and how these assays complement IHC and help guide therapy.
TREATMENT CARCINOMA OF UNKNOWN PRIMARY
GENERAL CONSIDERATIONS
The treatment of CUP continues to evolve, albeit slowly. The median survival duration of most patients with disseminated CUP is ~6–10 months. Systemic chemotherapy is the primary treatment modality in most patients with disseminated disease, but the careful integration of surgery, radiation therapy, and even periods of observation is important in the overall management of this condition (Figs. 120e-2 and 120e-3). Prognostic factors include performance status, site and number of metastases, response to chemotherapy, and serum lactate dehydrogenase (LDH) levels. Culine and colleagues developed a prognostic model using performance status and serum LDH levels, which allowed the assignment of patients into two subgroups with divergent outcomes. Future prospective trials using this prognostic model are warranted. Clinically, some CUP diagnoses fall into a favorable prognostic subset. Others, including those with disseminated CUP, do not and have a more unfavorable prognosis.
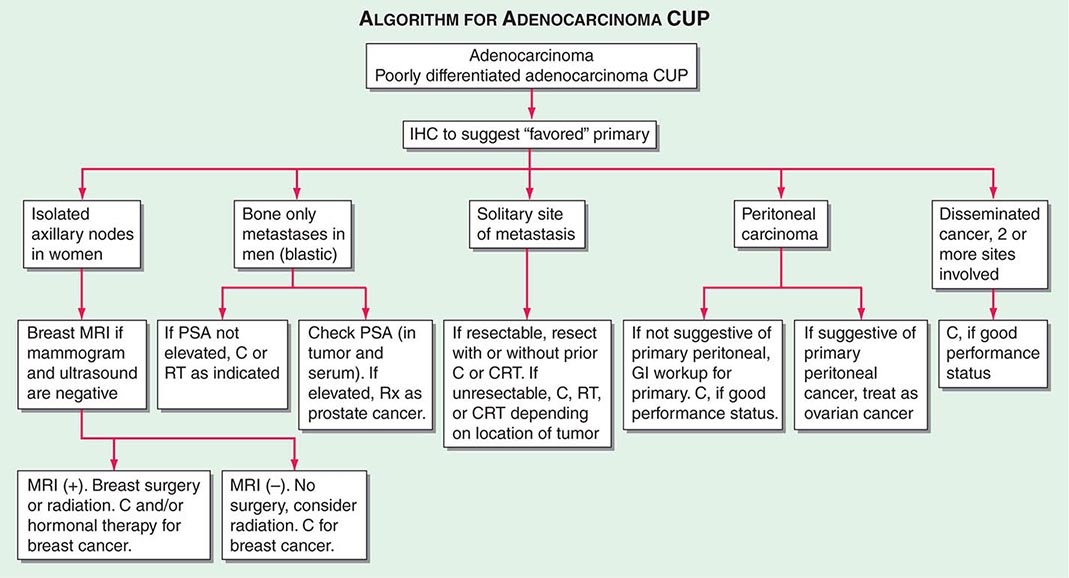
FIGURE 120e-2 Treatment algorithm for adenocarcinoma and poorly differentiated adenocarcinoma of unknown primary (CUP). C, chemotherapy; CRT, chemoradiation; GI, gastrointestinal; IHC, immunohistochemistry; MRI, magnetic resonance imaging; PSA, prostate-specific antigen; RT, radiation.
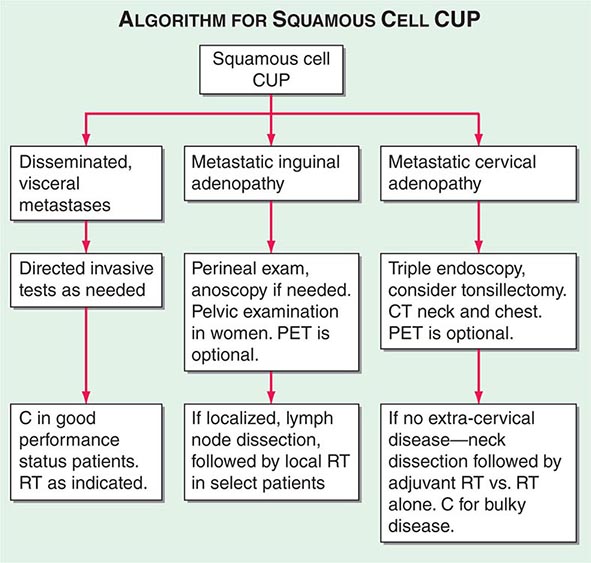
FIGURE 120e-3 Treatment algorithm for squamous cell carcinoma of unknown primary (CUP). C, chemotherapy; CT, computed tomography; PET, positron emission tomography; RT, radiation.
TREATMENT OF FAVORABLE CUP SUBSETS
Women with Isolated Axillary Adenopathy Women with isolated axillary adenopathy with adenocarcinoma or carcinoma are usually treated for stage II or III breast cancer based on pathologic findings. These patients should undergo a breast MRI if mammogram and ultrasound are negative. Radiation therapy to the ipsilateral breast is indicated if the breast MRI is positive. Chemotherapy and/or hormonal therapy are indicated based on patient’s age (premenopausal or postmenopausal), nodal disease bulk, and hormone receptor status (Chap. 108). It is important to verify that the pathology suggests a breast cancer profile (morphology, immunohistochemical breast markers including estrogen receptor, mammoglobin, GCDFP-15, HER-2 gene expression) before embarking on a breast cancer therapeutic program.
WOMEN WITH PERITONEAL CARCINOMATOSIS
The term primary peritoneal papillary serous carcinoma (PPSC) has been used to describe CUP with carcinomatosis with the pathologic and laboratory (elevated CA-125 antigen) characteristics of ovarian cancer but no ovarian primary tumor identified on transvaginal sonography or laparotomy. Studies suggest that ovarian cancer and PPSC, which are both of müllerian origin, have similar gene expression profiles. Similar to patients with ovarian cancer, patients with PPSC are candidates for cytoreductive surgery, followed by adjuvant taxane- and platinum-based chemotherapy. In one retrospective study of 258 women with peritoneal carcinomatosis who had undergone cytoreductive surgery and chemotherapy, 22% of patients had a complete response to chemotherapy; the median survival duration was 18 months (range 11–24 months). However, not all peritoneal carcinomatosis in women is PPSC. Careful pathologic evaluation can help diagnose a colon cancer profile (CDX-2+, CK-20+, CK7–) or a pancreaticobiliary cancer or even a mislabeled peritoneal mesothelioma (calretinin positive).
POORLY DIFFERENTIATED CARCINOMA WITH MIDLINE ADENOPATHY
Men with poorly differentiated or undifferentiated carcinoma that presents as a midline adenopathy should be evaluated for extragonadal germ cell malignancy. If diagnosed and treated as such, they often experience a good response to treatment with platinum-based combination chemotherapy. Response rates of >50% have been noted, and long-term survival rates of 10–15% long have been reported. Older patients (especially smokers) who present with mediastinal adenopathy are more likely to have a lung or head-and-neck cancer profile.
NEUROENDOCRINE CARCINOMA
Low-grade neuroendocrine carcinoma often has an indolent course, and treatment decisions are based on symptoms and tumor bulk. Urine 5-HIAA and serum chromogranin may be elevated and can be followed as markers. Often the patient is treated with somatostatin analogues alone for hormone-related symptoms (diarrhea, flushing, nausea). Specific local therapies or systemic therapy would only be indicated if the patient is symptomatic with local pain secondary to significant growth of the metastasis or the hormone-related symptoms are not controlled with endocrine therapy. Patients with high-grade neuroendocrine carcinoma are treated as having small-cell lung cancer and are responsive to chemotherapy; 20–25% show a complete response, and up to 10% patients survive more than 5 years.
SQUAMOUS CELL CARCINOMA PRESENTING AS NECK ADENOPATHY
Patients with early-stage squamous cell carcinoma involving the cervical lymph nodes are candidates for node dissection and radiation therapy, which can result in long-term survival. The role of chemotherapy in these patients is undefined, although chemoradiation therapy or induction chemotherapy is often used and is beneficial in bulky N2/N3 lymph node disease.
SOLITARY METASTATIC SITE
Patients with solitary metastases can also experience good treatment outcomes. Some patients who present with locoregional disease are candidates for aggressive trimodality management; both prolonged disease-free interval and occasionally cure are possible.
MEN WITH BLASTIC SKELETAL METASTASES AND ELEVATED PSA
Blastic bone-only metastasis is a rare presentation, and elevated serum PSA or tumor staining with PSA may provide confirmatory evidence of prostate cancer in these patients. Those with elevated levels are candidates for hormonal therapy for prostate cancer, although it is important to rule out other primary tumors (lung most common).
MANAGEMENT OF DISSEMINATED CUP
Patients who present with liver, brain, and adrenal metastatic disease usually have a poor prognosis. Patients with nonserous papillary primary peritoneal carcinomatosis can have a large differential diagnosis, which is mainly of gastrointestinal profile and includes gastric, appendiceal, colon, and pancreaticobiliary profiles.
Traditionally, platinum-based combination chemotherapy regimens have been used to treat CUP. Several broadly used regimens have been studied in the last two decades; these include paclitaxel-carboplatin, gemcitabine-cisplatin, gemcitabine-oxaliplatin, and irinotecan and fluoropyrimidine-based therapies. These chemotherapeutic agents used as empiric regimens have shown a response rate of 25–40%, and their use obtains median survival times of 6–13 months.
Outside of favorable subsets, there is a small group of patients with a “definitive” IHC. These patients usually have a single diagnosis based on their clinicopathologic presentation and are often treated for the putative primary tumor. This does not guarantee a response, although it increases the probability of response when select drugs are chosen from a class of drugs known to work in that cancer type. Patients who do not fall into those categories are candidates for broad-spectrum platinum-based regimens, clinical trials, and additional trial-based genomic and proteomic tests. Today, we do not have effective drugs for several CUP cancer profiles, and treatments overlap for some cancers. However, as novel therapies are developed for additional known cancers, tissue of origin and assessment of molecular features of the tumor will be important and might direct more selective treatment.
SUMMARY
Patients with CUP should undergo a directed diagnostic search for the primary tumor on the basis of clinical and pathologic data. Subsets of patients have prognostically favorable disease, as defined by clinical or histologic criteria, and may substantially benefit from aggressive treatment and expect prolonged survival. However, for most patients who present with advanced CUP, the prognosis remains poor with early resistance to available cytotoxic therapy. The current focus has shifted away from empirical chemotherapeutic trials to understanding the metastatic phenotype, tissue of origin profiling, and evaluating molecular targets in CUP patients.
121 | Paraneoplastic Syndromes: Endocrinologic/Hematologic |
Neoplastic cells can produce a variety of products that can stimulate hormonal, hematologic, dermatologic, and neurologic responses. Paraneoplastic syndromes is the term used to refer to the disorders that accompany benign or malignant tumors but are not directly related to mass effects or invasion. Tumors of neuroendocrine origin, such as small-cell lung carcinoma (SCLC) and carcinoids, produce a wide array of peptide hormones and are common causes of paraneoplastic syndromes. However, almost every type of tumor has the potential to produce hormones or to induce cytokine and immunologic responses. Careful studies of the prevalence of paraneoplastic syndromes indicate that they are more common than is generally appreciated. The signs, symptoms, and metabolic alterations associated with paraneoplastic disorders may be overlooked in the context of a malignancy and its treatment. Consequently, atypical clinical manifestations in a patient with cancer should prompt consideration of a paraneoplastic syndrome. The most common endocrinologic and hematologic syndromes associated with underlying neoplasia will be discussed here.
ENDOCRINE PARANEOPLASTIC SYNDROMES
Etiology Hormones can be produced from eutopic or ectopic sources. Eutopic refers to the expression of a hormone from its normal tissue of origin, whereas ectopic refers to hormone production from an atypical tissue source. For example, adrenocorticotropic hormone (ACTH) is expressed eutopically by the corticotrope cells of the anterior pituitary, but it can be expressed ectopically in SCLC. Many hormones are produced at low levels from a wide array of tissues in addition to the classic endocrine source. Thus, ectopic expression is often a quantitative change rather than an absolute change in tissue expression. Nevertheless, the term ectopic expression is firmly entrenched and conveys the abnormal physiology associated with hormone production by neoplastic cells. In addition to high levels of hormones, ectopic expression typically is characterized by abnormal regulation of hormone production (e.g., defective feedback control) and peptide processing (resulting in large, unprocessed precursors).
A diverse array of molecular mechanisms has been suggested to cause ectopic hormone production. In rare instances, genetic rearrangements explain aberrant hormone expression. For example, translocation of the parathyroid hormone (PTH) gene can result in high levels of PTH expression in tissues other than the parathyroid gland because the genetic rearrangement brings the PTH gene under the control of atypical regulatory elements. A related phenomenon is well documented in many forms of leukemia and lymphoma, in which somatic genetic rearrangements confer a growth advantage and alter cellular differentiation and function (Chap. 134). Although genetic rearrangements cause selected cases of ectopic hormone production, this mechanism is rare, as many tumors are associated with excessive production of numerous peptides. Cellular dedifferentiation probably underlies most cases of ectopic hormone production. Many cancers are poorly differentiated, and certain tumor products, such as human chorionic gonadotropin (hCG), parathyroid hormone–related protein (PTHrP), and α fetoprotein, are characteristic of gene expression at earlier developmental stages. In contrast, the propensity of certain cancers to produce particular hormones (e.g., squamous cell carcinomas produce PTHrP) suggests that dedifferentiation is partial or that selective pathways are derepressed. These expression profiles probably reflect epigenetic modifications that alter transcriptional repression, microRNA expression, and other pathways that govern cell differentiation.
In SCLC, the pathway of differentiation has been relatively well defined. The neuroendocrine phenotype is dictated in part by the basic-helix-loop-helix (bHLH) transcription factor human achaete-scute homologue 1 (hASH-1), which is expressed at abnormally high levels in SCLC associated with ectopic ACTH. The activity of hASH-1 is inhibited by hairy enhancer of split 1 (HES-1) and by Notch proteins, which also are capable of inducing growth arrest. Thus, abnormal expression of these developmental transcription factors appears to provide a link between cell proliferation and differentiation.
Ectopic hormone production would be merely an epiphenomenon associated with cancer if it did not result in clinical manifestations. Excessive and unregulated production of hormones such as ACTH, PTHrP, and vasopressin can lead to substantial morbidity and complicate the cancer treatment plan. Moreover, the paraneoplastic endocrinopathies may be a presenting clinical feature of underlying malignancy and prompt the search for an unrecognized tumor.
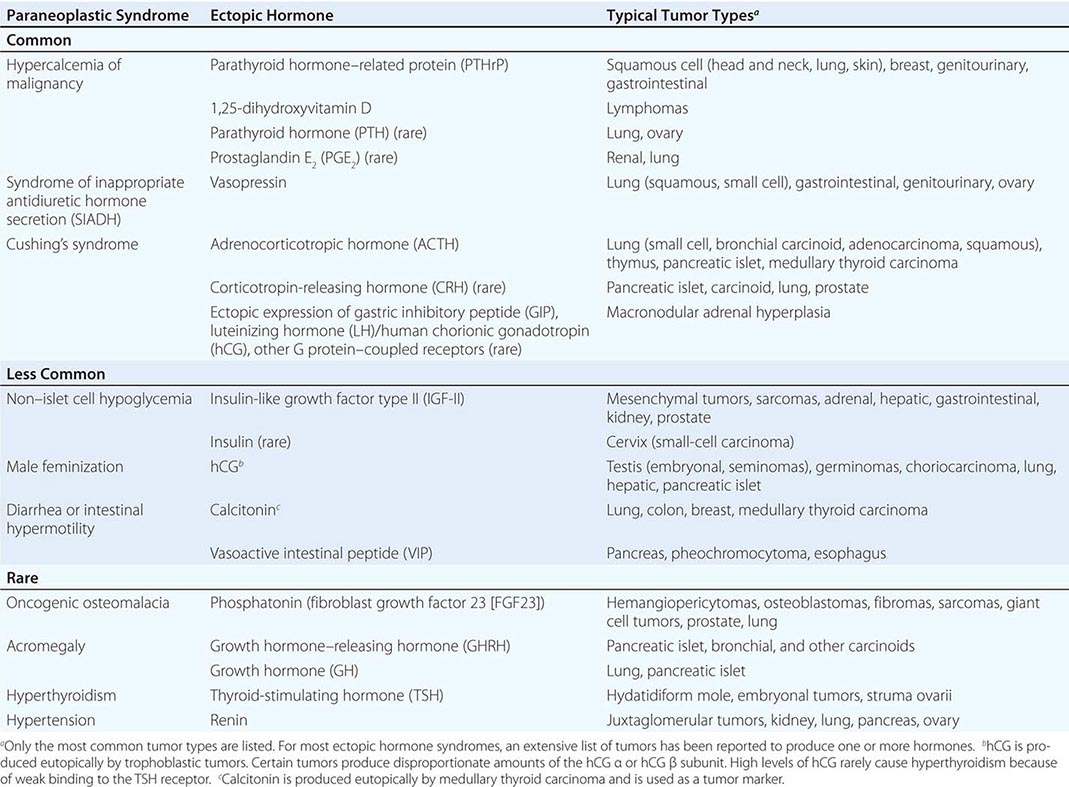
A large number of paraneoplastic endocrine syndromes have been described, linking overproduction of particular hormones with specific types of tumors. However, certain recurring syndromes emerge from this group (Table 121-1). The most common paraneoplastic endocrine syndromes include hypercalcemia from overproduction of PTHrP and other factors, hyponatremia from excess vasopressin, and Cushing’s syndrome from ectopic ACTH.
PARANEOPLASTIC SYNDROMES CAUSED BY ECTOPIC HORMONE PRODUCTION |
HYPERCALCEMIA CAUSED BY ECTOPIC PRODUCTION OF PTHrP
(See also Chap. 424)
Etiology Humoral hypercalcemia of malignancy (HHM) occurs in up to 20% of patients with cancer. HHM is most common in cancers of the lung, head and neck, skin, esophagus, breast, and genitourinary tract and in multiple myeloma and lymphomas. There are several distinct humoral causes of HHM, but it is caused most commonly by overproduction of PTHrP. In addition to acting as a circulating humoral factor, bone metastases (e.g., breast, multiple myeloma) may produce PTHrP, leading to local osteolysis and hypercalcemia. PTHrP may also affect the initiation and progression of tumors by acting through pro-survival and chemokine pathways.
PTHrP is structurally related to PTH and binds to the PTH receptor, explaining the similar biochemical features of HHM and hyperparathyroidism. PTHrP plays a key role in skeletal development and regulates cellular proliferation and differentiation in other tissues, including skin, bone marrow, breast, and hair follicles. The mechanism of PTHrP induction in malignancy is incompletely understood; however, tumor-bearing tissues commonly associated with HHM normally produce PTHrP during development or cell renewal. PTHrP expression is stimulated by hedgehog pathways and Gli transcription factors that are active in many malignancies. Transforming growth factor β (TGF-β), which is produced by many tumors, also stimulates PTHrP, in part by activating the Gli pathway. Mutations in certain oncogenes, such as Ras, also can activate PTHrP expression. In adult T cell lymphoma, the transactivating Tax protein produced by human T cell lymphotropic virus 1 (HTLV-1) stimulates PTHrP promoter activity. Metastatic lesions to bone are more likely to produce PTHrP than are metastases in other tissues, suggesting that bone produces factors (e.g., TGF-β) that enhance PTHrP production or that PTHrP-producing metastases have a selective growth advantage in bone. PTHrP activates the pro-survival AKT pathway and the chemokine receptor CXCR4. Thus, PTHrP production can be stimulated by mutations in oncogenes, altered expression of viral or cellular transcription factors, and local growth factors. In addition to its role in HHM, the PTHrP pathway may also provide a potential target for therapeutic intervention to impede cancer growth.
Another relatively common cause of HHM is excess production of 1,25-dihydroxyvitamin D. Like granulomatous disorders associated with hypercalcemia, lymphomas can produce an enzyme that converts 25-hydroxyvitamin D to the more active 1,25-dihydroxyvitamin D, leading to enhanced gastrointestinal calcium absorption. Other causes of HHM include tumor-mediated production of osteolytic cytokines and inflammatory mediators.
Clinical Manifestations The typical presentation of HHM is a patient with a known malignancy who is found to be hypercalcemic on routine laboratory tests. Less often, hypercalcemia is the initial presenting feature of malignancy. Particularly when calcium levels are markedly increased (>3.5 mmol/L [>14 mg/dL]), patients may experience fatigue, mental status changes, dehydration, or symptoms of nephrolithiasis.
Diagnosis Features that favor HHM, as opposed to primary hyperparathyroidism, include known malignancy, recent onset of hypercalcemia, and very high serum calcium levels. Like hyperparathyroidism, hypercalcemia caused by PTHrP is accompanied by hypercalciuria and hypophosphatemia. Patients with HHM typically have metabolic alkalosis rather than hyperchloremic acidosis, as is seen in hyperparathyroidism. Measurement of PTH is useful to exclude primary hyperparathyroidism; the PTH level should be suppressed in HHM. An elevated PTHrP level confirms the diagnosis, and it is increased in ~80% of hypercalcemic patients with cancer. 1,25-Dihydroxyvitamin D levels may be increased in patients with lymphoma.
ECTOPIC VASOPRESSIN: TUMOR-ASSOCIATED SIADH
(See also Chap. 63)
Etiology Vasopressin is an antidiuretic hormone normally produced by the posterior pituitary gland. Ectopic vasopressin production by tumors is a common cause of the syndrome of inappropriate antidiuretic hormone (SIADH), occurring in at least half of patients with SCLC. SIADH also can be caused by a number of nonneoplastic conditions, including central nervous system (CNS) trauma, infections, and medications (Chap. 404). Compensatory responses to SIADH, such as decreased thirst, may mitigate the development of hyponatremia. However, with prolonged production of excessive vasopressin, the osmostat controlling thirst and hypothalamic vasopressin secretion may become reset. In addition, intake of free water, orally or intravenously, can quickly worsen hyponatremia because of reduced renal diuresis.
Tumors with neuroendocrine features, such as SCLC and carcinoids, are the most common sources of ectopic vasopressin production, but it also occurs in other forms of lung cancer and with CNS lesions, head and neck cancer, and genitourinary, gastrointestinal, and ovarian cancers. The mechanism of activation of the vasopressin gene in these tumors is unknown but often involves concomitant expression of the adjacent oxytocin gene, suggesting derepression of this locus.
Clinical Manifestations Most patients with ectopic vasopressin secretion are asymptomatic and are identified because of the presence of hyponatremia on routine chemistry testing. Symptoms may include weakness, lethargy, nausea, confusion, depressed mental status, and seizures. The severity of symptoms reflects the rapidity of onset as well as the severity of hyponatremia. Hyponatremia usually develops slowly but may be exacerbated by the administration of IV fluids or the institution of new medications.
Diagnosis The diagnostic features of ectopic vasopressin production are the same as those of other causes of SIADH (Chaps. 63 and 404). Hyponatremia and reduced serum osmolality occur in the setting of an inappropriately normal or increased urine osmolality. Urine sodium excretion is normal or increased unless volume depletion is present. Other causes of hyponatremia should be excluded, including renal, adrenal, or thyroid insufficiency. Physiologic sources of vasopressin stimulation (CNS lesions, pulmonary disease, nausea), adaptive circulatory mechanisms (hypotension, heart failure, hepatic cirrhosis), and medications, including many chemotherapeutic agents, also should be considered as possible causes of hyponatremia. Vasopressin measurements are not usually necessary to make the diagnosis.
CUSHING’S SYNDROME CAUSED BY ECTOPIC ACTH PRODUCTION
(See also Chap. 406)
Etiology Ectopic ACTH production accounts for 10–20% of cases of Cushing’s syndrome. The syndrome is particularly common in neuroendocrine tumors. SCLC is the most common cause of ectopic ACTH, followed by bronchial and thymic carcinoids, islet cell tumors, other carcinoids, and pheochromocytomas. Ectopic ACTH production is caused by increased expression of the proopiomelanocortin (POMC) gene, which encodes ACTH, along with melanocyte-stimulating hormone (MSH), β lipotropin, and several other peptides. In many tumors, there is abundant but aberrant expression of the POMC gene from an internal promoter, proximal to the third exon, which encodes ACTH. However, because this product lacks the signal sequence necessary for protein processing, it is not secreted. Increased production of ACTH arises instead from less abundant, but unregulated, POMC expression from the same promoter site used in the pituitary. However, because the tumors lack many of the enzymes needed to process the POMC polypeptide, it is typically released as multiple large, biologically inactive fragments along with relatively small amounts of fully processed, active ACTH.
Rarely, corticotropin-releasing hormone (CRH) is produced by pancreatic islet cell tumors, SCLC, medullary thyroid cancer, carcinoids, or prostate cancer. When levels are high enough, CRH can cause pituitary corticotrope hyperplasia and Cushing’s syndrome. Tumors that produce CRH sometimes also produce ACTH, raising the possibility of a paracrine mechanism for ACTH production.
A distinct mechanism for ACTH-independent Cushing’s syndrome involves ectopic expression of various G protein–coupled receptors in the adrenal nodules. Ectopic expression of the gastric inhibitory peptide (GIP) receptor is the best-characterized example of this mechanism. In this case, meals induce GIP secretion, which inappropriately stimulates adrenal growth and glucocorticoid production.
Clinical Manifestations The clinical features of hypercortisolemia are detected in only a small fraction of patients with documented ectopic ACTH production. Patients with ectopic ACTH syndrome generally exhibit less marked weight gain and centripetal fat redistribution, probably because the exposure to excess glucocorticoids is relatively brief and because cachexia reduces the propensity for weight gain and fat deposition. The ectopic ACTH syndrome is associated with several clinical features that distinguish it from other causes of Cushing’s syndrome (e.g., pituitary adenomas, adrenal adenomas, iatrogenic glucocorticoid excess). The metabolic manifestations of ectopic ACTH syndrome are dominated by fluid retention and hypertension, hypokalemia, metabolic alkalosis, glucose intolerance, and occasionally steroid psychosis. The very high ACTH levels often cause increased pigmentation, and melanotrope-stimulating hormone (MSH) activity derived from the POMC precursor peptide is also increased. The extraordinarily high glucocorticoid levels in patients with ectopic sources of ACTH can lead to marked skin fragility and easy bruising. In addition, the high cortisol levels often overwhelm the renal 11β-hydroxysteroid dehydrogenase type II enzyme, which normally inactivates cortisol and prevents it from binding to renal mineralocorticoid receptors. Consequently, in addition to the excess mineralocorticoids produced by ACTH stimulation of the adrenal gland, high levels of cortisol exert activity through the mineralocorticoid receptor, leading to severe hypokalemia.
Diagnosis The diagnosis of ectopic ACTH syndrome is usually not difficult in the setting of a known malignancy. Urine free cortisol levels fluctuate but are typically greater than two to four times normal, and the plasma ACTH level is usually >22 pmol/L (>100 pg/mL). A suppressed ACTH level excludes this diagnosis and indicates an ACTH-independent cause of Cushing’s syndrome (e.g., adrenal or exogenous glucocorticoid). In contrast to pituitary sources of ACTH, most ectopic sources of ACTH do not respond to glucocorticoid suppression. Therefore, high-dose dexamethasone (8 mg PO) suppresses 8:00 A.M. serum cortisol (50% decrease from baseline) in ~80% of pituitary ACTH-producing adenomas but fails to suppress ectopic ACTH in ~90% of cases. Bronchial and other carcinoids are well-documented exceptions to these general guidelines, as these ectopic sources of ACTH may exhibit feedback regulation indistinguishable from pituitary adenomas, including suppression by high-dose dexamethasone, and ACTH responsiveness to adrenal blockade with metyrapone. If necessary, petrosal sinus catheterization can be used to evaluate a patient with ACTH-dependent Cushing’s syndrome when the source of ACTH is unclear. After CRH stimulation, a 3:1 petrosal sinus:peripheral ACTH ratio strongly suggests a pituitary ACTH source. Imaging studies (computed tomography or magnetic resonance imaging) are also useful in the evaluation of suspected carcinoid lesions, allowing biopsy and characterization of hormone production using special stains. If available, positron emission tomography or octreotide scanning may identify some sources of ACTH production.
TUMOR-INDUCED HYPOGLYCEMIA CAUSED BY EXCESS PRODUCTION OF IGF-II
(See also Chap. 420) Mesenchymal tumors, hemangiopericytomas, hepatocellular tumors, adrenal carcinomas, and a variety of other large tumors have been reported to produce excessive amounts of insulin-like growth factor type II (IGF-II) precursor, which binds weakly to insulin receptors and more strongly to IGF-I receptors, leading to insulin-like actions. The gene encoding IGF-II resides on a chromosome 11p15 locus that is normally imprinted (that is, expression is exclusively from a single parental allele). Biallelic expression of the IGF-II gene occurs in a subset of tumors, suggesting loss of methylation and loss of imprinting as a mechanism for gene induction. In addition to increased IGF-II production, IGF-II bioavailability is increased due to complex alterations in circulating binding proteins. Increased IGF-II suppresses growth hormone (GH) and insulin, resulting in reduced IGF binding protein 3 (IGFBP-3), IGF-I, and acid-labile subunit (ALS). The reduction in ALS and IGFBP-3, which normally sequester IGF-II, causes it to be displaced to a small circulating complex that has greater access to insulin target tissues. For this reason, circulating IGF-II levels may not be markedly increased despite causing hypoglycemia. In addition to IGF-II–mediated hypoglycemia, tumors may occupy enough of the liver to impair gluconeogenesis.
In most cases, a tumor causing hypoglycemia is clinically apparent (usually >10 cm in size) and hypoglycemia develops in association with fasting. The diagnosis is made by documenting low serum glucose and suppressed insulin levels in association with symptoms of hypoglycemia. Serum IGF-II levels may not be increased (IGF-II assays may not detect IGF-II precursors). Increased IGF-II mRNA expression is found in most of these tumors. Any medications associated with hypoglycemia should be eliminated. Treatment of the underlying malignancy, if possible, may reduce the predisposition to hypoglycemia. Frequent meals and IV glucose, especially during sleep or fasting, are often necessary to prevent hypoglycemia. Glucagon and glucocorticoids have also been used to enhance glucose production.
HUMAN CHORIONIC GONADOTROPIN
hCG is composed of α and β subunits and can be produced as intact hormone, which is biologically active, or as uncombined biologically inert subunits. Ectopic production of intact hCG occurs most often in association with testicular embryonal tumors, germ cell tumors, extragonadal germinomas, lung cancer, hepatoma, and pancreatic islet tumors. Eutopic production of hCG occurs with trophoblastic malignancies. hCG α subunit production is particularly common in lung cancer and pancreatic islet cancer. In men, high hCG levels stimulate steroidogenesis and aromatase activity in testicular Leydig cells, resulting in increased estrogen production and the development of gynecomastia. Precocious puberty in boys or gynecomastia in men should prompt measurement of hCG and consideration of a testicular tumor or another source of ectopic hCG production. Most women are asymptomatic. hCG is easily measured. Treatment should be directed at the underlying malignancy.
ONCOGENIC OSTEOMALACIA
Hypophosphatemic oncogenic osteomalacia, also called tumor-induced osteomalacia (TIO), is characterized by markedly reduced serum phosphorus and renal phosphate wasting, leading to muscle weakness, bone pain, and osteomalacia. Serum calcium and PTH levels are normal, and 1,25-dihydroxyvitamin D is low. Oncogenic osteomalacia is usually caused by benign mesenchymal tumors, such as hemangiopericytomas, fibromas, and giant cell tumors, often of the skeletal extremities or head. It has also been described in sarcomas and in patients with prostate and lung cancer. Resection of the tumor reverses the disorder, confirming its humoral basis. The circulating phosphaturic factor is called phosphatonin—a factor that inhibits renal tubular reabsorption of phosphate and renal conversion of 25-hydroxyvitamin D to 1,25-dihydroxyvitamin D. Phosphatonin has been identified as fibroblast growth factor 23 (FGF23). FGF23 levels are increased in some, but not all, patients with osteogenic osteomalacia. FGF23 forms a ternary complex with the klotho protein and renal FGF receptors to reduce renal phosphate reabsorption. Treatment involves removal of the tumor, if possible, and supplementation with phosphate and vitamin D. Octreotide treatment reduces phosphate wasting in some patients with tumors that express somatostatin receptor subtype 2. Octreotide scans may also be useful in detecting these tumors.
HEMATOLOGIC SYNDROMES
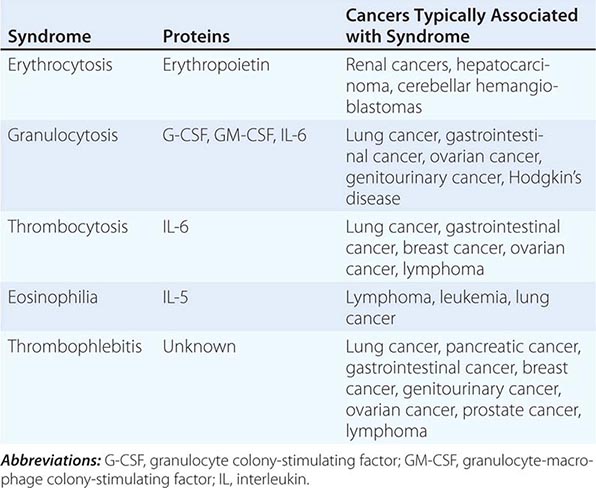
The elevation of granulocyte, platelet, and eosinophil counts in most patients with myeloproliferative disorders is caused by the proliferation of the myeloid elements due to the underlying disease rather than to a paraneoplastic syndrome. The paraneoplastic hematologic syndromes in patients with solid tumors are less well characterized than are the endocrine syndromes because the ectopic hormone(s) or cytokines responsible have not been identified in most of these tumors (Table 121-2). The extent of the paraneoplastic syndromes parallels the course of the cancer.
PARANEOPLASTIC HEMATOLOGIC SYNDROMES |
ERYTHROCYTOSIS
Ectopic production of erythropoietin by cancer cells causes most paraneoplastic erythrocytosis. The ectopically produced erythropoietin stimulates the production of red blood cells (RBCs) in the bone marrow and raises the hematocrit. Other lymphokines and hormones produced by cancer cells may stimulate erythropoietin release but have not been proved to cause erythrocytosis.
Most patients with erythrocytosis have an elevated hematocrit (>52% in men, >48% in women) that is detected on a routine blood count. Approximately 3% of patients with renal cell cancer, 10% of patients with hepatoma, and 15% of patients with cerebellar hemangioblastomas have erythrocytosis. In most cases, the erythrocytosis is asymptomatic.
Patients with erythrocytosis due to a renal cell cancer, hepatoma, or CNS cancer should have measurement of red cell mass. If the red cell mass is elevated, the serum erythropoietin level should be measured. Patients with an appropriate cancer, elevated erythropoietin levels, and no other explanation for erythrocytosis (e.g., hemoglobinopathy that causes increased O2 affinity; Chap. 77) have the paraneoplastic syndrome.
GRANULOCYTOSIS
Approximately 30% of patients with solid tumors have granulocytosis (granulocyte count >8000/μL). In about half of patients with granulocytosis and cancer, the granulocytosis has an identifiable nonparaneoplastic etiology (infection, tumor necrosis, glucocorticoid administration, etc.). The other patients have proteins in urine and serum that stimulate the growth of bone marrow cells. Tumors and tumor cell lines from patients with lung, ovarian, and bladder cancers have been documented to produce granulocyte colony-stimulating factor (G-CSF), granulocyte-macrophage colony-stimulating factor (GM-CSF), and/or interleukin 6 (IL-6). However, the etiology of granulocytosis has not been characterized in most patients.
Patients with granulocytosis are nearly all asymptomatic, and the differential white blood cell count does not have a shift to immature forms of neutrophils. Granulocytosis occurs in 40% of patients with lung and gastrointestinal cancers, 20% of patients with breast cancer, 30% of patients with brain tumors and ovarian cancers, 20% of patients with Hodgkin’s disease, and 10% of patients with renal cell carcinoma. Patients with advanced-stage disease are more likely to have granulocytosis than are those with early-stage disease.
Paraneoplastic granulocytosis does not require treatment. The granulocytosis resolves when the underlying cancer is treated.
THROMBOCYTOSIS
Some 35% of patients with thrombocytosis (platelet count >400,000/μL) have an underlying diagnosis of cancer. IL-6, a candidate molecule for the etiology of paraneoplastic thrombocytosis, stimulates the production of platelets in vitro and in vivo. Some patients with cancer and thrombocytosis have elevated levels of IL-6 in plasma. Another candidate molecule is thrombopoietin, a peptide hormone that stimulates megakaryocyte proliferation and platelet production. The etiology of thrombocytosis has not been established in most cases.
Patients with thrombocytosis are nearly all asymptomatic. Thrombocytosis is not clearly linked to thrombosis in patients with cancer. Thrombocytosis is present in 40% of patients with lung and gastrointestinal cancers; 20% of patients with breast, endometrial, and ovarian cancers; and 10% of patients with lymphoma. Patients with thrombocytosis are more likely to have advanced-stage disease and have a poorer prognosis than do patients without thrombocytosis. In ovarian cancer, IL-6 has been shown to directly promote tumor growth. Paraneoplastic thrombocytosis does not require treatment other than treatment of the underlying tumor.
EOSINOPHILIA
Eosinophilia is present in ~1% of patients with cancer. Tumors and tumor cell lines from patients with lymphomas or leukemia may produce IL-5, which stimulates eosinophil growth. Activation of IL-5 transcription in lymphomas and leukemias may involve translocation of the long arm of chromosome 5, to which the genes for IL-5 and other cytokines map.
Patients with eosinophilia are typically asymptomatic. Eosinophilia is present in 10% of patients with lymphoma, 3% of patients with lung cancer, and occasional patients with cervical, gastrointestinal, renal, and breast cancer. Patients with markedly elevated eosinophil counts (>5000/μL) can develop shortness of breath and wheezing. A chest radiograph may reveal diffuse pulmonary infiltrates from eosinophil infiltration and activation in the lungs.
THROMBOPHLEBITIS
Deep venous thrombosis and pulmonary embolism are the most common thrombotic conditions in patients with cancer. Migratory or recurrent thrombophlebitis may be the initial manifestation of cancer. Nearly 15% of patients who develop deep venous thrombosis or pulmonary embolism have a diagnosis of cancer (Chap. 142). The coexistence of peripheral venous thrombosis with visceral carcinoma, particularly pancreatic cancer, is called Trousseau’s syndrome.
Pathogenesis Patients with cancer are predisposed to thromboembolism because they are often at bed rest or immobilized, and tumors may obstruct or slow blood flow. Postoperative deep venous thrombosis is twice as common in cancer patients who undergo surgery. Chronic IV catheters also predispose to clotting. In addition, clotting may be promoted by release of procoagulants or cytokines from tumor cells or associated inflammatory cells or by platelet adhesion or aggregation. The specific molecules that promote thromboembolism have not been identified.
Chemotherapeutic agents, particularly those associated with endothelial damage, can induce venous thrombosis. The annual risk of venous thrombosis in patients with cancer receiving chemotherapy is about 11%, sixfold higher than the risk in the general population. Bleomycin, L-asparaginase, thalidomide analogues, cisplatin-based regimens, and high doses of busulfan and carmustine are all associated with an increased risk.
In addition to cancer and its treatment causing secondary thrombosis, primary thrombophilic diseases may be associated with cancer. For example, the antiphospholipid antibody syndrome is associated with a wide range of pathologic manifestations (Chap. 379). About 20% of patients with this syndrome have cancers. Among patients with cancer and antiphospholipid antibodies, 35–45% develop thrombosis.
Clinical Manifestations Patients with cancer who develop deep venous thrombosis usually develop swelling or pain in the leg, and physical examination reveals tenderness, warmth, and redness. Patients who present with pulmonary embolism develop dyspnea, chest pain, and syncope, and physical examination shows tachycardia, cyanosis, and hypotension. Some 5% of patients with no history of cancer who have a diagnosis of deep venous thrombosis or pulmonary embolism will have a diagnosis of cancer within 1 year. The most common cancers associated with thromboembolic episodes include lung, pancreatic, gastrointestinal, breast, ovarian, and genitourinary cancers; lymphomas; and brain tumors. Patients with cancer who undergo surgical procedures requiring general anesthesia have a 20–30% risk of deep venous thrombosis.
Diagnosis The diagnosis of deep venous thrombosis in patients with cancer is made by impedance plethysmography or bilateral compression ultrasonography of the leg veins. Patients with a noncompressible venous segment have deep venous thrombosis. If compression ultrasonography is normal and there is a high clinical suspicion for deep venous thrombosis, venography should be done to look for a luminal filling defect. Elevation of D-dimer is not as predictive of deep venous thrombosis in patients with cancer as it is in patients without cancer; elevations are seen in people over age 65 years without concomitant evidence of thrombosis, probably as a consequence of increased thrombin deposition and turnover in aging.
Patients with symptoms and signs suggesting a pulmonary embolism should be evaluated with a chest radiograph, electrocardiogram, arterial blood gas analysis, and ventilation-perfusion scan. Patients with mismatched segmental perfusion defects have a pulmonary embolus. Patients with equivocal ventilation-perfusion findings should be evaluated as described above for deep venous thrombosis in their legs. If deep venous thrombosis is detected, they should be anticoagulated. If deep venous thrombosis is not detected, they should be considered for a pulmonary angiogram.
Patients without a diagnosis of cancer who present with an initial episode of thrombophlebitis or pulmonary embolus need no additional tests for cancer other than a careful history and physical examination. In light of the many possible primary sites, diagnostic testing in asymptomatic patients is wasteful. However, if the clot is refractory to standard treatment or is in an unusual site or if the thrombophlebitis is migratory or recurrent, efforts to find an underlying cancer are indicated.
Cutaneous paraneoplastic syndromes are discussed in Chap. 72. Neurologic paraneoplastic syndromes are discussed in Chap. 122.
ACKNOWLEDGMENT
The authors acknowledge the contributions of Bruce E. Johnson to prior versions of this chapter.
122 | Paraneoplastic Neurologic Syndromes and Autoimmune Encephalitis |
Paraneoplastic neurologic disorders (PNDs) are cancer-related syndromes that can affect any part of the nervous system (Table 122-1). They are caused by mechanisms other than metastasis or by any of the complications of cancer such as coagulopathy, stroke, metabolic and nutritional conditions, infections, and side effects of cancer therapy. In 60% of patients, the neurologic symptoms precede the cancer diagnosis. Clinically disabling PNDs occur in 0.5–1% of all cancer patients, but they affect 2–3% of patients with neuroblastoma or small-cell lung cancer (SCLC) and 30–50% of patients with thymoma or sclerotic myeloma.
PARANEOPLASTIC SYNDROMES OF THE NERVOUS SYSTEM |

PATHOGENESIS
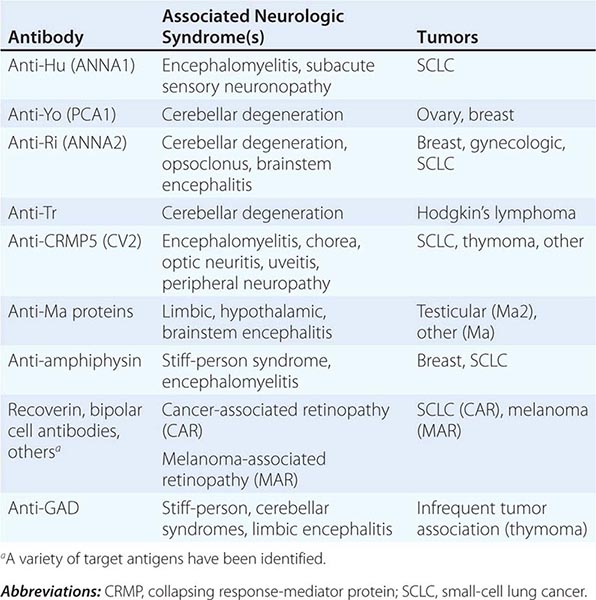
Most PNDs are mediated by immune responses triggered by neuronal proteins (onconeuronal antigens) expressed by tumors. In PNDs of the central nervous system (CNS), many antibody-associated immune responses have been identified (Table 122-2). These antibodies react with the patient’s tumor, and their detection in serum or cerebrospinal fluid (CSF) usually predicts the presence of cancer. When the antigens are intracellular, most syndromes are associated with extensive infiltrates of CD4+ and CD8+ T cells, microglial activation, gliosis, and variable neuronal loss. The infiltrating T cells are often in close contact with neurons undergoing degeneration, suggesting a primary pathogenic role. T cell–mediated cytotoxicity may contribute directly to cell death in these PNDs. Thus both humoral and cellular immune mechanisms participate in the pathogenesis of many PNDs. This complex immunopathogenesis may underlie the resistance of many of these conditions to therapy.
ANTIBODIES TO INTRACELLULAR ANTIGENS, SYNDROMES, AND ASSOCIATED CANCERS |
In contrast to the disorders associated with immune responses against intracellular antigens, those associated with antibodies to antigens expressed on the neuronal cell surface of the CNS or at the neuromuscular junction are more responsive to immunotherapy (Table 122-3, Fig. 122-1). These disorders occur with and without a cancer association and may affect children and young adults, and there is increasing evidence that they are mediated by the antibodies.
ANTIBODIES TO CELL SURFACE OR SYNAPTIC ANTIGENS, SYNDROMES, AND ASSOCIATED TUMORS |
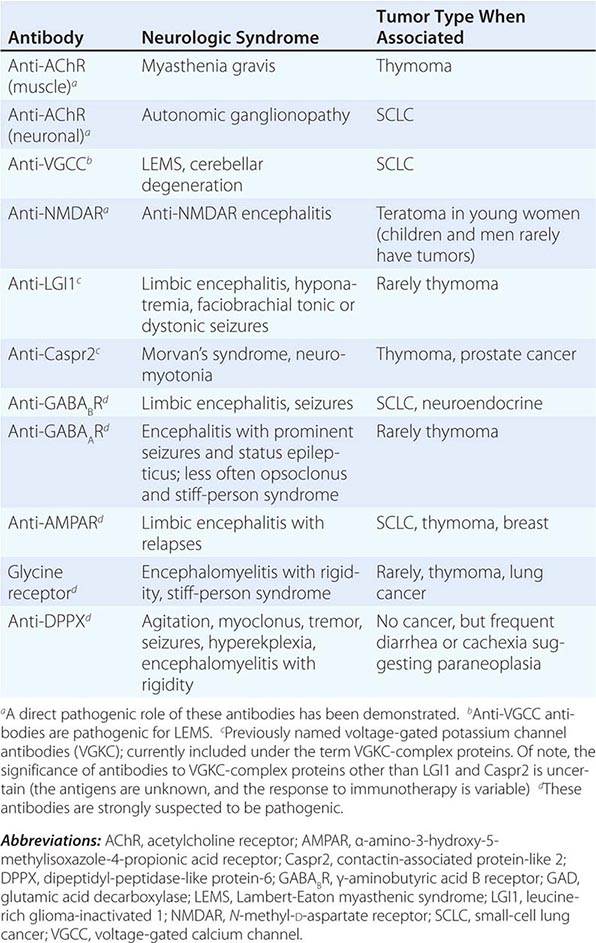
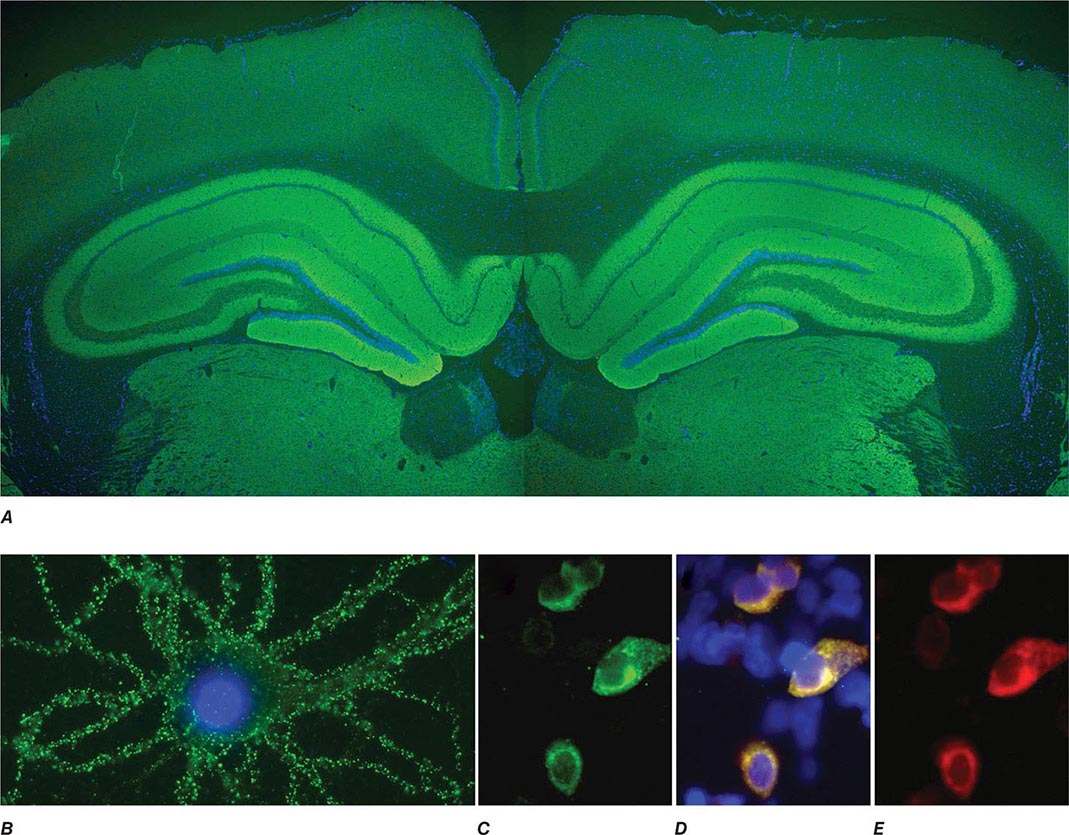
FIGURE 122-1 Antibodies to the GluN1 subunit of the N-methyl-D-aspartate (NMDA) receptor in a patient with anti-NMDA receptor encephalitis and ovarian teratoma. (A) Coronal section of rat brain immunolabeled (green fluorescence) with the patient’s antibodies. The reactivity predominates in the hippocampus, which is highly enriched in NMDA receptors. (B) This image shows the antibody reactivity with cultures of rat hippocampal neurons; the intense green immunolabeling is due to the antibodies against the GluN1 subunit of NMDA receptors. (C–E) Images of HEK cells (a human kidney cell line) transfected to express NMDA receptors, showing reactivity with patient’s antibodies (C) and with a commercial monoclonal antibody against NMDA receptors (E); the patient’s antibody reactivity co-labels only the cells that express NMDA receptors (D). (From J Dalmau et al: Lancet Neurol 7:1091, 2008; with permission.)
Other PNDs are likely immune-mediated, although their antigens are unknown. These include several syndromes of inflammatory neuropathies and myopathies. In addition, many patients with typical PND syndromes are antibody-negative.
For still other PNDs, the cause remains quite obscure. These include, among others, several neuropathies that occur in the terminal stages of cancer and a number of neuropathies associated with plasma cell dyscrasias or lymphoma without evidence of inflammatory infiltrates or deposits of immunoglobulin, cryoglobulin, or amyloid.
APPROACH TO THE PATIENT:
Paraneoplastic Neurologic Disorders
Three key concepts are important for the diagnosis and management of PNDs. First, it is common for symptoms to appear before the presence of a tumor is known; second, the neurologic syndrome usually develops rapidly, producing severe deficits in a short period of time; and third, there is evidence that prompt tumor control improves the neurologic outcome. Therefore, the major concern of the physician is to recognize a disorder promptly as paraneoplastic and to identify and treat the tumor.
PND OF THE CENTRAL NERVOUS SYSTEM AND DORSAL ROOT GANGLIA
When symptoms involve brain, spinal cord, or dorsal root ganglia, the suspicion of PND is usually based on a combination of clinical, radiologic, and CSF findings. Presence of antineuronal antibodies (Tables 122-2 and 122-3) may help in the diagnosis, but only 60–70% of PNDs of the CNS and less than 20% of those involving the peripheral nervous system have neuronal or neuromuscular junction antibodies that can be used as diagnostic tests.
Magnetic resonance imaging (MRI) and CSF studies are important to rule out neurologic complications due to the direct spread of cancer, particularly metastatic and leptomeningeal disease. In most PNDs, the MRI findings are nonspecific. Paraneoplastic limbic encephalitis is usually associated with characteristic MRI abnormalities in the mesial temporal lobes (see below), but similar findings can occur with other disorders (e.g., nonparaneoplastic autoimmune limbic encephalitis and human herpesvirus type 6 [HHV-6] encephalitis) (Fig. 122-2). The CSF profile of patients with PND of the CNS or dorsal root ganglia typically consists of mild to moderate pleocytosis (<200 mononuclear cells, predominantly lymphocytes), an increase in the protein concentration, and a variable presence of oligoclonal bands. There are no specific electrophysiologic tests that are diagnostic of PND. Moreover, a biopsy of the affected tissue is often difficult to obtain, and although useful to rule out other disorders (e.g., metastasis) the pathologic findings are not specific for PND.
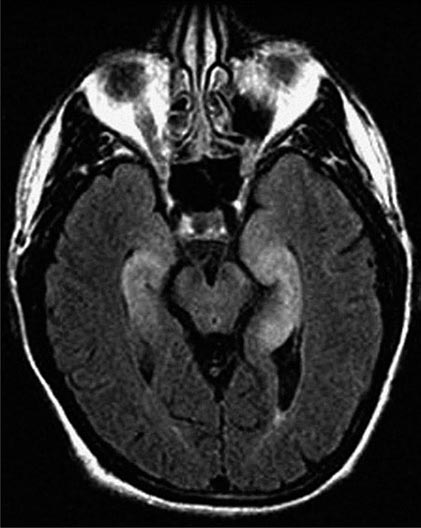
FIGURE 122-2 Fluid-attenuated inversion recovery sequence magnetic resonance imaging of a patient with limbic encephalitis and LGI1 antibodies. Note the abnormal hyperintensity involving the medial aspect of the temporal lobes.
PND OF NERVE AND MUSCLE
If symptoms involve peripheral nerve, neuromuscular junction, or muscle, the diagnosis of a specific PND is usually established on clinical, electrophysiologic, and pathologic grounds. The clinical history, accompanying symptoms (e.g., anorexia, weight loss), and type of syndrome dictate the studies and degree of effort needed to demonstrate a neoplasm. For example, the frequent association of Lambert-Eaton myasthenic syndrome (LEMS) with SCLC should lead to a chest and abdomen computed tomography (CT) or body positron emission tomography (PET) scan and, if negative, periodic tumor screening for at least 3 years after the neurologic diagnosis. In contrast, the weak association of polymyositis with cancer calls into question the need for repeated cancer screenings in this situation. Serum and urine immunofixation studies should be considered in patients with peripheral neuropathy of unknown cause; detection of a monoclonal gammopathy suggests the need for additional studies to uncover a B cell or plasma cell malignancy. In paraneoplastic neuropathies, diagnostically useful antineuronal antibodies are limited to anti-CV2/CRMP5 and anti-Hu.
For any type of PND, if antineuronal antibodies are negative, the diagnosis relies on the demonstration of cancer and the exclusion of other cancer-related or independent neurologic disorders. Combined CT and PET scans often uncover tumors undetected by other tests. For germ cell tumors of the testis and teratomas of the ovary, ultrasound and CT or MRI of the abdomen and pelvis may reveal tumors undetectable by PET.
SPECIFIC PARANEOPLASTIC NEUROLOGIC SYNDROMES
PARANEOPLASTIC ENCEPHALOMYELITIS AND FOCAL ENCEPHALITIS
The term encephalomyelitis describes an inflammatory process with multifocal involvement of the nervous system, including brain, brainstem, cerebellum, and spinal cord. It is often associated with dorsal root ganglia and autonomic dysfunction. For any given patient, the clinical manifestations are determined by the areas predominantly involved, but pathologic studies almost always reveal abnormalities beyond the symptomatic regions. Several clinicopathologic syndromes may occur alone or in combination: (1) cortical encephalitis, which may present as “epilepsia partialis continua”; (2) limbic encephalitis, characterized by confusion, depression, agitation, anxiety, severe short-term memory deficits, partial complex seizures, and sometimes dementia (the MRI usually shows unilateral or bilateral medial temporal lobe abnormalities, best seen with T2 and fluid-attenuated inversion recovery sequences); (3) brainstem encephalitis, resulting in eye movement disorders (nystagmus, opsoclonus, supranuclear or nuclear paresis), cranial nerve paresis, dysarthria, dysphagia, and central autonomic dysfunction; (4) cerebellar gait and limb ataxia; (5) myelitis, which may cause lower or upper motor neuron symptoms, myoclonus, muscle rigidity, and spasms; and (6) autonomic dysfunction as a result of involvement of the neuraxis at multiple levels, including hypothalamus, brainstem, and autonomic nerves (see Paraneoplastic Peripheral Neuropathies, below). Cardiac arrhythmias, postural hypotension, and central hypoventilation are frequent causes of death in patients with encephalomyelitis.
Paraneoplastic encephalomyelitis and focal encephalitis are usually associated with SCLC, but many other cancers have been implicated. Patients with SCLC and these syndromes usually have anti-Hu antibodies in serum and CSF. Anti-CRMP5 antibodies occur less frequently; some of these patients may develop chorea, uveitis, or optic neuritis. Antibodies to Ma proteins are associated with limbic, hypothalamic, and brainstem encephalitis and occasionally with cerebellar symptoms (Fig. 122-3); some patients develop hypersomnia, cataplexy, and severe hypokinesia. MRI abnormalities are frequent, including those described with limbic encephalitis and variable involvement of the hypothalamus, basal ganglia, or upper brainstem. The oncologic associations of these antibodies are shown in Table 122-2.
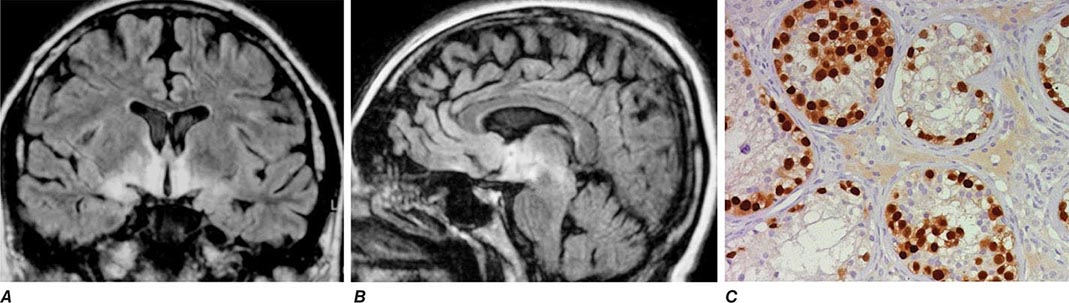
FIGURE 122-3 Magnetic resonance imaging (MRI) and tumor of a patient with anti-Ma2-associated encephalitis. (A and B) Fluid-attenuated inversion recovery MRI sequences showing abnormal hyperintensities in the medial temporal lobes, hypothalamus, and upper brainstem. (C) This image corresponds to a section of the patient’s orchiectomy incubated with a specific marker (Oct4) of germ cell tumors. The positive (brown) cells correspond to an intratubular germ cell neoplasm.
ENCEPHALITIDES WITH ANTIBODIES TO CELL-SURFACE OR SYNAPTIC PROTEINS (TABLE 122-3)
These disorders are important for three reasons: (1) they can occur with and without tumor association, (2) some syndromes predominate in young individuals and children, and (3) despite the severity of the symptoms patients usually respond to treatment of the tumor, if found, and immunotherapy (e.g., glucocorticoids, IVIg, plasma exchange, rituximab, or cyclophosphamide).
Encephalitis with N-methyl-D-aspartate (NMDA) receptor antibodies (Fig. 122-1) usually occurs in young women and children, but men and older patients of both sexes can be affected. The disorder has a characteristic pattern of symptom progression that includes a prodrome resembling a viral process, followed in a few days by the onset of severe psychiatric symptoms, memory loss, seizures, decreased level of consciousness, abnormal movements (orofacial, limb, and trunk dyskinesias, dystonic postures), autonomic instability, and frequent hypoventilation. Monosymptomatic episodes, such as pure psychosis, occur in 4% of the patients. Clinical relapses occur in 12–24% of patients (12% during the first 2 years after initial presentation). Most patients have intrathecal synthesis of antibodies, likely by infiltrating plasma cells in brain and meninges (Fig. 122-4A). The syndrome is often misdiagnosed as a viral or idiopathic encephalitis, neuroleptic malignant syndrome, or encephalitis lethargica, and many patients are initially evaluated by psychiatrists with the suspicion of acute psychosis. The detection of an associated teratoma is dependent on age and gender: 46% of female patients 12 years or older have uni- or bilateral ovarian teratomas, whereas less than 7% of girls younger than 12 have a teratoma (Fig. 122-4B). In male patients, the detection of a tumor is rare. Patients older than 45 years are more frequently male; about 20% of these patients have tumors (e.g., cancer of the breast, ovary, or lung).
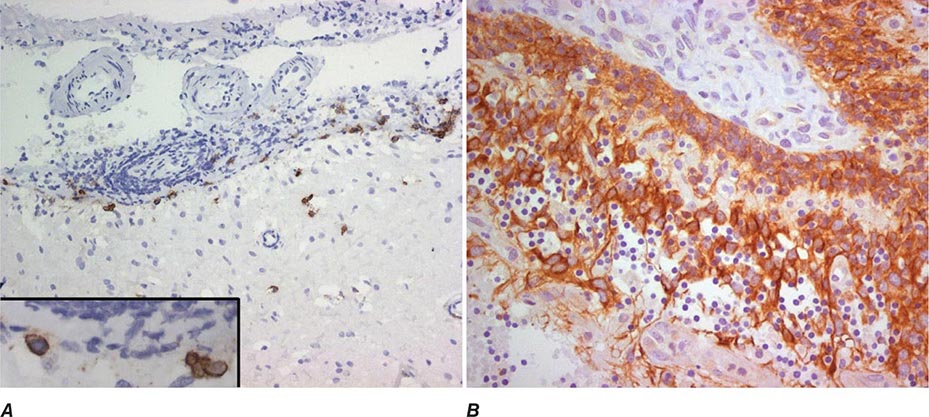
FIGURE 122-4 Pathologic findings in anti–N-methyl-D-aspartate (NMDA) receptor encephalitis. Infiltrates of plasma cells (brown cells; stained for CD138) in the meninges and brain of a patient (A); the inset is a magnification of some plasma cells. (B) Neurons and neuronal processes in the teratoma of a patient (brown cells; stained with MAP2); these neurons express NMDA receptors (not shown). (From E Martinez-Hernandez et al: Neurology 77:589, 2011, with permission.)
Encephalitis with leucine-rich glioma-inactivated 1 (LGI1) antibodies predominates in patients older than 50 years (65% male) and frequently presents with memory loss and seizures (limbic encephalopathy), along with hyponatremia and sleep dysfunction. In a small number of patients, the encephalitis is preceded by or occurs with myoclonic-like movements called faciobrachial dystonic or tonic seizures. Less than 10% of patients have thymoma.
Encephalitis with contactin-associated protein-like 2 (Caspr2) antibodies predominates in patients older than 50 years and is associated with Morvan’s syndrome (encephalitis, insomnia, confusion, hallucinations, autonomic dysfunction, and neuromyotonia) and, less frequently, with limbic encephalitis, neuromyotonia, and neuropathic pain. About 30–40% of patients have thymoma.
Encephalitis with γ-aminobutyric acid type B (GABAB) receptor antibodies is usually associated with limbic encephalitis and seizures. In rare instances, patients develop cerebellar symptoms and opsoclonus. Fifty percent of patients have SCLC or a neuroendocrine tumor of the lung. Patients may have additional antibodies to glutamic acid decarboxylase (GAD), which are of unclear significance. Other antibodies to nonneuronal proteins are often found in these patients as well as in patients with α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) receptor antibodies, indicating a general tendency to autoimmunity.
Encephalitis with GABAA receptor antibodies may affect children and adults. When antibodies are present at high titer in serum and CSF, the disorder associates with prominent seizures and status epilepticus, often requiring pharmacologically induced coma. Low titer antibodies in serum are often associated with other autoimmune conditions, and the spectrum of symptoms is wider, including encephalitis, seizures, opsoclonus, or stiff-person syndrome. Most patients do not have an underlying tumor, but some may have thymoma.
Encephalitis with AMPA receptor antibodies affects middle-aged women, who develop acute limbic dysfunction or, less frequently, prominent psychiatric symptoms; 70% of the patients have an underlying tumor in the lung, breast, or thymus. Neurologic relapses may occur; these also respond to immunotherapy and are not necessarily associated with tumor recurrence.
Encephalitis with glycine receptor (GlyR) antibodies has been described in adults with progressive encephalomyelitis with rigidity and myoclonus (PERM) and stiff-person spectrum of symptoms (with or without GAD antibodies). The disorder usually occurs without tumor association, although some patients have lung cancer, thymoma, or Hodgkin’s lymphoma.
Encephalitis with dipeptidyl-peptidase-like protein-6 (or DPPX) antibodies results in symptoms of CNS hyperexcitability including agitation, hallucinations, paranoid delusions, tremor, myoclonus, nystagmus, seizures, and sometimes hyperekplexia. Some patients develop progressive encephalomyelitis with rigidity and myoclonus. Diarrhea, other gastrointestinal symptoms, and substantial loss of weight often suggest the presence of an underlying tumor, but no tumor association has been identified. The disorder responds to immunotherapy.
PARANEOPLASTIC CEREBELLAR DEGENERATION
This disorder is often preceded by a prodrome that may include dizziness, oscillopsia, blurry or double vision, nausea, and vomiting. A few days or weeks later, patients develop dysarthria, gait and limb ataxia, and variable dysphagia. The examination usually shows downbeating nystagmus and, rarely, opsoclonus. Brainstem dysfunction, upgoing toes, or a mild neuropathy may occur. Early in the course, MRI studies are usually normal; later, the MRI reveals cerebellar atrophy. The disorder results from extensive degeneration of Purkinje cells, with variable involvement of other cerebellar cortical neurons, deep cerebellar nuclei, and spinocerebellar tracts. The tumors more frequently involved are SCLC, cancer of the breast and ovary, and Hodgkin’s lymphoma.
Anti-Yo antibodies in patients with breast and gynecologic cancers and anti-Tr antibodies in patients with Hodgkin’s lymphoma are the two immune responses typically associated with prominent or pure cerebellar degeneration. Antibodies to P/Q-type voltage-gated calcium channels (VGCC) occur in some patients with SCLC and cerebellar dysfunction; only some of these patients develop LEMS. A variable degree of cerebellar dysfunction can be associated with virtually any of the antibodies and PND of the CNS shown in Table 122-2.
A number of single case reports have described neurologic improvement after tumor removal, plasma exchange, IVIg, cyclophosphamide, rituximab, or glucocorticoids. However, most patients with paraneoplastic cerebellar degeneration do not improve with treatment.
PARANEOPLASTIC OPSOCLONUS-MYOCLONUS SYNDROME
Opsoclonus is a disorder of eye movement characterized by involuntary, chaotic saccades that occur in all directions of gaze; it is frequently associated with myoclonus and ataxia. Opsoclonus-myoclonus may be cancer-related or idiopathic. When the cause is paraneoplastic, the tumors involved are usually cancer of the lung and breast in adults, neuroblastoma in children, and ovarian teratoma in adolescents and young women. The pathologic substrate of opsoclonus-myoclonus is unclear, but studies suggest that disinhibition of the fastigial nucleus of the cerebellum is involved. Most patients do not have antineuronal antibodies. A small subset of patients with ataxia, opsoclonus, and other eye-movement disorders develop anti-Ri antibodies; in rare instances, muscle rigidity, laryngeal spasms, autonomic dysfunction, and dementia also occur. The tumors most frequently involved in anti-Ri-associated syndromes are breast and ovarian cancer. If the tumor is not successfully treated, the syndrome in adults often progresses to encephalopathy, coma, and death. In addition to treating the tumor, symptoms may respond to immunotherapy (glucocorticoids, plasma exchange, and/or IVIg).
At least 50% of children with opsoclonus-myoclonus have an underlying neuroblastoma. Hypotonia, ataxia, behavioral changes, and irritability are frequent accompanying symptoms. Neurologic symptoms often improve with treatment of the tumor and glucocorticoids, adrenocorticotropic hormone (ACTH), plasma exchange, IVIg, and rituximab. Many patients are left with psychomotor retardation and behavioral and sleep problems.
PARANEOPLASTIC SYNDROMES OF THE SPINAL CORD
The number of reports of paraneoplastic spinal cord syndromes, such as subacute motor neuronopathy and acute necrotizing myelopathy, has decreased in recent years. This may represent a true decrease in incidence, due to improved and prompt oncologic interventions, or the identification of nonparaneoplastic etiologies. Some patients with cancer develop upper or lower motor neuron dysfunction or both, resembling amyotrophic lateral sclerosis. It is unclear whether these disorders have a paraneoplastic etiology or simply coincide with the presence of cancer. There are isolated case reports of cancer patients with motor neuron dysfunction who had neurologic improvement after tumor treatment. A search for lymphoma should be undertaken in patients with a rapidly progressive motor neuron syndrome and a monoclonal protein in serum or CSF.
Paraneoplastic myelitis may present with upper or lower motor neuron symptoms, segmental myoclonus, and rigidity, and can be the first manifestation of encephalomyelitis. Neuromyelitis optica (NMO) with aquaporin 4 antibodies may occur in rare instances as a paraneoplastic manifestation of a cancer.
PARANEOPLASTIC STIFF-PERSON SYNDROME
This disorder is characterized by progressive muscle rigidity, stiffness, and painful spasms triggered by auditory, sensory, or emotional stimuli. Rigidity mainly involves the lower trunk and legs, but it can affect the upper extremities and neck. Sometimes, only one extremity is affected (stiff-limb syndrome). Symptoms improve with sleep and general anesthetics. Electrophysiologic studies demonstrate continuous motor unit activity. The associated antibodies target proteins (GAD, amphiphysin) involved in the function of inhibitory synapses using γ-aminobutyric acid (GABA) or glycine as neurotransmitters. The presence of amphiphysin antibodies usually indicates a paraneoplastic etiology related to SCLC and breast cancer. By contrast, GAD antibodies may occur in some cancer patients but are much more frequently present in the nonparaneoplastic disorder. GlyR antibodies may occur in some patients with stiff-person syndrome; these antibodies are also detectable in patients with PERM.
Optimal treatment of stiff-person syndrome requires therapy of the underlying tumor, glucocorticoids, and symptomatic use of drugs that enhance GABA-ergic transmission (diazepam, baclofen, sodium valproate, tiagabine, vigabatrin). IVIg and plasma exchange are transiently effective in some patients.
PARANEOPLASTIC SENSORY NEURONOPATHY OR DORSAL ROOT GANGLIONOPATHY
This syndrome is characterized by sensory deficits that may be symmetric or asymmetric, painful dysesthesias, radicular pain, and decreased or absent reflexes. All modalities of sensation and any part of the body including face and trunk can be involved. Specialized sensations such as taste and hearing can also be affected. Electrophysiologic studies show decreased or absent sensory nerve potentials with normal or near-normal motor conduction velocities. Symptoms result from an inflammatory, likely immune-mediated, process that targets the dorsal root ganglia, causing neuronal loss and secondary degeneration of the posterior columns of the spinal cord. The dorsal and, less frequently, the anterior nerve roots and peripheral nerves may also be involved. This disorder often precedes or is associated with encephalomyelitis and autonomic dysfunction and has the same immunologic and oncologic associations (Hu antibodies, SCLC).
As with anti-Hu-associated encephalomyelitis, the therapeutic approach focuses on prompt treatment of the tumor. Glucocorticoids occasionally produce clinical stabilization or improvement. The benefit of IVIg and plasma exchange is not proven.
PARANEOPLASTIC PERIPHERAL NEUROPATHIES
These disorders may develop any time during the course of the neoplastic disease. Neuropathies occurring at late stages of cancer or lymphoma usually cause mild to moderate sensorimotor deficits due to axonal degeneration of unclear etiology. These neuropathies are often masked by concurrent neurotoxicity from chemotherapy and other cancer therapies. In contrast, the neuropathies that develop in the early stages of cancer frequently show a rapid progression, sometimes with a relapsing and remitting course, and evidence of inflammatory infiltrates and axonal loss or demyelination. If demyelinating features predominate (Chaps. 459 and 460), IVIg, plasma exchange, or glucocorticoids may improve symptoms. Occasionally anti-CRMP5 antibodies are present; detection of anti-Hu suggests concurrent dorsal root ganglionitis.
Guillain-Barré syndrome and brachial plexitis have occasionally been reported in patients with lymphoma, but there is no clear evidence of a paraneoplastic association (Chap. 460).
Malignant monoclonal gammopathies include: (1) multiple myeloma and sclerotic myeloma associated with IgG or IgA monoclonal proteins; and (2) Waldenström’s macroglobulinemia, B cell lymphoma, and chronic B cell lymphocytic leukemia associated with IgM monoclonal proteins. These disorders may cause neuropathy by a variety of mechanisms, including compression of roots and plexuses by metastasis to vertebral bodies and pelvis, deposits of amyloid in peripheral nerves, and paraneoplastic mechanisms. The paraneoplastic variety has several distinctive features. Approximately half of patients with sclerotic myeloma develop a sensorimotor neuropathy with predominantly motor deficits, resembling a chronic inflammatory demyelinating neuropathy (Chap. 460); some patients develop elements of the POEMS syndrome (polyneuropathy, organomegaly, endocrinopathy, M protein, skin changes). Treatment of the plasmacytoma or sclerotic lesions usually improves the neuropathy. In contrast, the sensorimotor or sensory neuropathy associated with multiple myeloma is more refractory to treatment. Between 5 and 10% of patients with Waldenström’s macroglobulinemia develop a distal symmetric sensorimotor neuropathy with predominant involvement of large sensory fibers. These patients may have IgM antibodies in their serum against myelin-associated glycoprotein and various gangliosides (Chap. 460). In addition to treating the Waldenström’s macroglobulinemia, other therapies may improve the neuropathy, including plasma exchange, IVIg, chlorambucil, cyclophosphamide, fludarabine, or rituximab.
Vasculitis of the nerve and muscle causes a painful symmetric or asymmetric distal axonal sensorimotor neuropathy with variable proximal weakness. It predominantly affects elderly men and is associated with an elevated erythrocyte sedimentation rate and increased CSF protein concentration. SCLC and lymphoma are the primary tumors involved. Glucocorticoids and cyclophosphamide often result in neurologic improvement.
Peripheral nerve hyperexcitability (neuromyotonia, or Isaacs’ syndrome) is characterized by spontaneous and continuous muscle fiber activity of peripheral nerve origin. Clinical features include cramps, muscle twitching (fasciculations or myokymia), stiffness, delayed muscle relaxation (pseudomyotonia), and spontaneous or evoked carpal or pedal spasms. The involved muscles may be hypertrophic, and some patients develop paresthesias and hyperhidrosis. CNS dysfunction, including mood changes, sleep disorder, hallucinations, and autonomic symptoms may occur. The electromyogram (EMG) shows fibrillations; fasciculations; and doublet, triplet, or multiplet single-unit (myokymic) discharges that have a high intraburst frequency. Some patients have Caspr2 antibodies in the context of Morvan’s syndrome, but most cases of isolated neuromyotonia are antibody negative. The disorder often occurs without cancer; if paraneoplastic, benign and malignant thymomas and SCLC are the usual tumors. Phenytoin, carbamazepine, and plasma exchange improve symptoms.
Paraneoplastic autonomic neuropathy usually develops as a component of other disorders, such as LEMS and encephalomyelitis. It may rarely occur as a pure or predominantly autonomic neuropathy with cholinergic or adrenergic dysfunction at the pre- or postganglionic levels. Patients can develop several life-threatening complications, such as gastrointestinal paresis with pseudo-obstruction, cardiac dysrhythmias, and postural hypotension. Other clinical features include abnormal pupillary responses, dry mouth, anhidrosis, erectile dysfunction, and problems in sphincter control. The disorder occurs in association with several tumors, including SCLC, cancer of the pancreas or testis, carcinoid tumors, and lymphoma. Because autonomic symptoms can be the presenting feature of encephalomyelitis, serum anti-Hu and anti-CRMP5 antibodies should be sought. Antibodies to ganglionic (alpha3-type) neuronal acetylcholine receptors are the cause of autoimmune autonomic ganglionopathy, a disorder that frequently occurs without cancer association (Chap. 454).
LAMBERT-EATON MYASTHENIC SYNDROME
LEMS is discussed in Chap. 461.
MYASTHENIA GRAVIS
Myasthenia gravis is discussed in Chap. 461.
POLYMYOSITIS-DERMATOMYOSITIS
Polymyositis and dermatomyositis are discussed in detail in Chap. 388.
ACUTE NECROTIZING MYOPATHY
Patients with this syndrome develop myalgias and rapid progression of weakness involving the extremities and the pharyngeal and respiratory muscles, often resulting in death. Serum muscle enzymes are elevated, and muscle biopsy shows extensive necrosis with minimal or absent inflammation and sometimes deposits of complement. The disorder occurs as a paraneoplastic manifestation of a variety of cancers including SCLC and cancer of the gastrointestinal tract, breast, kidney, and prostate, among others. Glucocorticoids and treatment of the underlying tumor rarely control the disorder.
PARANEOPLASTIC VISUAL SYNDROMES
This group of disorders involves the retina and, less frequently, the uvea and optic nerves. The term cancer-associated retinopathy is used to describe paraneoplastic cone and rod dysfunction characterized by photosensitivity, progressive loss of vision and color perception, central or ring scotomas, night blindness, and attenuation of photopic and scotopic responses in the electroretinogram (ERG). The most commonly associated tumor is SCLC. Melanoma-associated retinopathy affects patients with metastatic cutaneous melanoma. Patients develop acute onset of night blindness and shimmering, flickering, or pulsating photopsias that often progress to visual loss. The ERG shows reduced b waves with normal dark adapted a waves. Paraneoplastic optic neuritis and uveitis are very uncommon and can develop in association with encephalomyelitis. Some patients with paraneoplastic uveitis and optic neuritis have anti-CRMP5 antibodies.
Some paraneoplastic retinopathies are associated with serum antibodies that specifically react with the subset of retinal cells undergoing degeneration, supporting an immune-mediated pathogenesis (Table 122-2). Paraneoplastic retinopathies usually fail to improve with treatment, although rare responses to glucocorticoids, plasma exchange, and IVIg have been reported.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


