Invasive Fungal Infections
KEY CONCEPTS
![]() Systemic mycoses can be caused by pathogenic fungi and include histoplasmosis, coccidioidomycosis, cryptococcosis, blastomycosis, paracoccidioidomycosis, and sporotrichosis, or infections by opportunistic fungi such as Candida albicans, Aspergillus species, Trichosporon, Candida glabrata, Fusarium, Alternaria, and Mucor.
Systemic mycoses can be caused by pathogenic fungi and include histoplasmosis, coccidioidomycosis, cryptococcosis, blastomycosis, paracoccidioidomycosis, and sporotrichosis, or infections by opportunistic fungi such as Candida albicans, Aspergillus species, Trichosporon, Candida glabrata, Fusarium, Alternaria, and Mucor.
![]() The diagnosis of fungal infection generally is accomplished by careful evaluation of clinical symptoms, results of serologic tests, and histopathologic examination and culture of clinical specimens.
The diagnosis of fungal infection generally is accomplished by careful evaluation of clinical symptoms, results of serologic tests, and histopathologic examination and culture of clinical specimens.
![]() Histoplasmosis is caused by Histoplasma capsulatum and is endemic in parts of the central United States along the Ohio and Mississippi River valleys. Although most patients experience asymptomatic infection, some can experience chronic, disseminated disease.
Histoplasmosis is caused by Histoplasma capsulatum and is endemic in parts of the central United States along the Ohio and Mississippi River valleys. Although most patients experience asymptomatic infection, some can experience chronic, disseminated disease.
![]() Asymptomatic patients with histoplasmosis are not treated, although patients who do not have acquired immune deficiency syndrome (AIDS) patients with evident disease are treated with either oral ketoconazole or IV amphotericin B; AIDS patients are treated with amphotericin B and then receive lifelong suppression.
Asymptomatic patients with histoplasmosis are not treated, although patients who do not have acquired immune deficiency syndrome (AIDS) patients with evident disease are treated with either oral ketoconazole or IV amphotericin B; AIDS patients are treated with amphotericin B and then receive lifelong suppression.
![]() Blastomycosis is caused by Blastomyces dermatitidis. In the immunocompetent host, acute pulmonary blastomycosis can be mild and self-limited and may not require treatment. However, consideration should be given to treating all infected individuals to prevent extrapulmonary dissemination. All persons with moderate to severe pneumonia, disseminated infection, or those who are immunocompromised require antifungal therapy.
Blastomycosis is caused by Blastomyces dermatitidis. In the immunocompetent host, acute pulmonary blastomycosis can be mild and self-limited and may not require treatment. However, consideration should be given to treating all infected individuals to prevent extrapulmonary dissemination. All persons with moderate to severe pneumonia, disseminated infection, or those who are immunocompromised require antifungal therapy.
![]() Coccidioidomycosis is caused by Coccidioides immitis and is endemic in some parts of the southwestern United States. It can cause nonspecific symptoms, acute pneumonia, or chronic pulmonary or disseminated disease. Primary pulmonary disease (unless severe) frequently is not treated, whereas extrapulmonary disease is treated with amphotericin B, and meningitis is treated with fluconazole.
Coccidioidomycosis is caused by Coccidioides immitis and is endemic in some parts of the southwestern United States. It can cause nonspecific symptoms, acute pneumonia, or chronic pulmonary or disseminated disease. Primary pulmonary disease (unless severe) frequently is not treated, whereas extrapulmonary disease is treated with amphotericin B, and meningitis is treated with fluconazole.
![]() Cryptococcosis is caused by Cryptococcus neoformans, which occurs primarily in immunocompromised patients, and Cryptococcus gattii, which occurs primarily in nonimmunocompromised patients. Patients with acute meningitis are treated with amphotericin B with flucytosine. Patients infected with human immunodeficiency virus (HIV) often require long-term suppressive therapy with fluconazole or itraconazole.
Cryptococcosis is caused by Cryptococcus neoformans, which occurs primarily in immunocompromised patients, and Cryptococcus gattii, which occurs primarily in nonimmunocompromised patients. Patients with acute meningitis are treated with amphotericin B with flucytosine. Patients infected with human immunodeficiency virus (HIV) often require long-term suppressive therapy with fluconazole or itraconazole.
![]() A variety of Candida species (including C. albicans, C. glabrata, Candida tropicalis, Candida parapsilosis, and Candida krusei) can cause diseases such as mucocutaneous, oral, esophageal, vaginal, and hematogenous candidiasis, as well as candiduria. Candidemia can be treated with a variety of antifungal agents; the optimal choice depends on previous patient exposure to antifungal agents, potential drug interactions and toxicities of each agent, and local epidemiology of intensive care unit (ICU) or hematology–oncology centers.
A variety of Candida species (including C. albicans, C. glabrata, Candida tropicalis, Candida parapsilosis, and Candida krusei) can cause diseases such as mucocutaneous, oral, esophageal, vaginal, and hematogenous candidiasis, as well as candiduria. Candidemia can be treated with a variety of antifungal agents; the optimal choice depends on previous patient exposure to antifungal agents, potential drug interactions and toxicities of each agent, and local epidemiology of intensive care unit (ICU) or hematology–oncology centers.
![]() Aspergillosis can be caused by a variety of Aspergillus species that can cause superficial infections, pneumonia, allergic bronchopulmonary aspergillosis (BPA), or invasive infection. Voriconazole has emerged as the drug of choice of most clinicians for primary therapy of most patients with invasive aspergillosis (IA). Combination therapy, while widely used, lacks clinical trial data to support its use.
Aspergillosis can be caused by a variety of Aspergillus species that can cause superficial infections, pneumonia, allergic bronchopulmonary aspergillosis (BPA), or invasive infection. Voriconazole has emerged as the drug of choice of most clinicians for primary therapy of most patients with invasive aspergillosis (IA). Combination therapy, while widely used, lacks clinical trial data to support its use.
For many years, fungal infections were classified as either superficial “nuisance diseases,” such as athlete’s foot or vulvovaginal candidiasis, or as relatively rare infections confined primarily to endemic areas of the country. When invasive fungal infections were encountered, amphotericin B was the only consistently effective, systemically active agent available for the treatment of systemic mycoses. ![]() Advances in medical technology including organ and bone marrow transplantation, cytotoxic chemotherapy, the widespread use of indwelling IV catheters, and the increased use of potent broad-spectrum antimicrobial agents all have contributed to the dramatic increase in the incidence of fungal infections worldwide.
Advances in medical technology including organ and bone marrow transplantation, cytotoxic chemotherapy, the widespread use of indwelling IV catheters, and the increased use of potent broad-spectrum antimicrobial agents all have contributed to the dramatic increase in the incidence of fungal infections worldwide.
Fungal infections have emerged as a major cause of death among cancer patients and transplant recipients.1–3 In addition, patients with acquired immune deficiency syndrome (AIDS) experience substantially more frequent and severe forms of cryptococcosis, histoplasmosis, coccidioidomycosis, and mucocutaneous (esophageal, oral, and vulvovaginal) candidiasis.
Problems remain in the diagnosis, prevention, and treatment of fungal infections. Unlike the available diagnostic techniques for most bacterial pathogens, there remains a host of unresolved issues regarding standardization of susceptibility testing methods, in vitro and in vivo models of infection, the usefulness of monitoring antifungal plasma concentrations, and the development and identification of resistant pathogens.1,4–6 The Infectious Diseases Society of America (IDSA) publishes guidelines for the treatment of many commonly encountered fungal infections.7–12 These guidelines provide summaries of the literature and a consensus of expert opinions regarding the treatment of these difficult infections.
MYCOLOGY
Fungi are eukaryotic organisms with a defined nucleus enclosed by a nuclear membrane; a cytoplasmic membrane containing lipids, glycoproteins, and sterols, mitochondria, Golgi apparatus, and ribosomes bound to endoplasmic reticulum; and a cytoskeleton with microtubules, microfilaments, and intermediate filaments. Fungi have rigid cell walls composed of chitin, cellulose, or both that stain with Gomori methenamine silver or periodic acid–Schiff reagent. Most fungi, except Candida species, are too weakly gram-positive to be seen well on Gram stain. Cryptococcus neoformans has a polysaccharide capsule surrounding the cell wall.1
Morphologically, pathogenic fungi can be grouped as either filamentous molds or unicellular yeasts (Fig. 99–1). Molds grow as multicellular branching, threadlike filaments (hyphae) that are either septate (divided by transverse walls) or coenocytic (multinucleate without cross walls). On agar media, molds grow outward from the point of inoculation by extension of the tips of filaments and then branch repeatedly, interweaving to form fuzzy, matted growths called mycelia. Yeasts are oval or spherically shaped unicellular forms that generally produce pasty or mucoid colonies on agar medium similar to those observed with bacterial cultures. Yeasts have rigid cell walls and reproduce by budding, a process in which daughter cells arise from pinching off a portion of the parent cell.
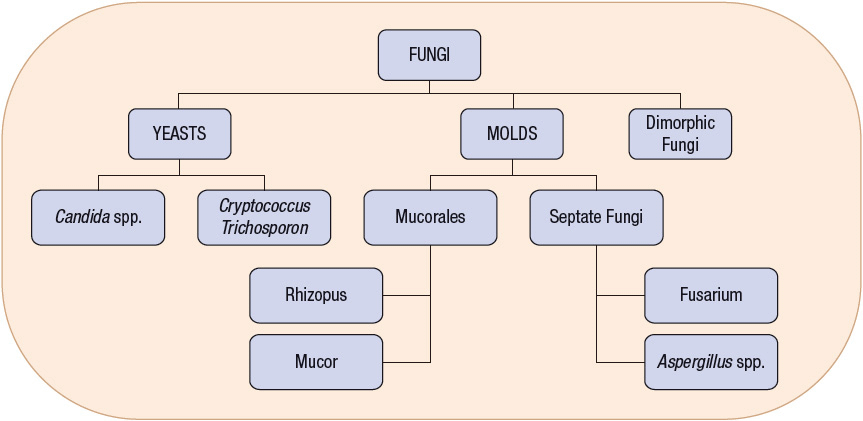
FIGURE 99-1 Morphologically, pathogenic fungi can be grouped as either filamentous molds or unicellular yeasts. Molds grow as multicellular branching, thread-like filaments (hyphae) that are either septate (divided by transverse walls) or coenocytic (multinucleate without cross walls).
Many pathogenic fungi, termed dimorphic fungi, exist as either a yeast or a mold, depending on pathogen, site of growth (in the host or in the laboratory setting), and temperature. Usually yeasts are the parasitic form that invades human or animal host tissue, whereas molds are the free-living form found in the environment. For example, Histoplasma capsulatum exists as a yeast in humans and as a mold in the laboratory.1
Susceptibility Testing of Antifungal Agents
Most laboratories do not routinely perform susceptibility tests on fungal isolates, but standardized methods for performing these tests are being developed and are now available for testing selected yeasts.
The Clinical and Laboratory Standards Institute (CLSI) defined clinical breakpoints (CBPs) for fluconazole, itraconazole, voriconazole, and flucytosine for all Candida species. Breakpoints are antimicrobial concentrations (MICs) obtained from susceptibility testing, which are used to define isolates as susceptible, intermediate, or resistant. No CBPs have been established for posaconazole or amphotericin B versus Candida.6 (Tables 99–1 and 99-2). Reliable and convincing interpretive breakpoints are not yet available for amphotericin B since available methodology does not reliably identify amphotericin B-resistant isolates.5–7 The breakpoints should be used following testing with the standardized, reproducible laboratory methodology used to develop the test and they should be interpreted in the context of the delivered dose of the antifungal agent.
TABLE 99-1 General Patterns of Susceptibility and Interpretive Breakpoints of Candida Speciesa
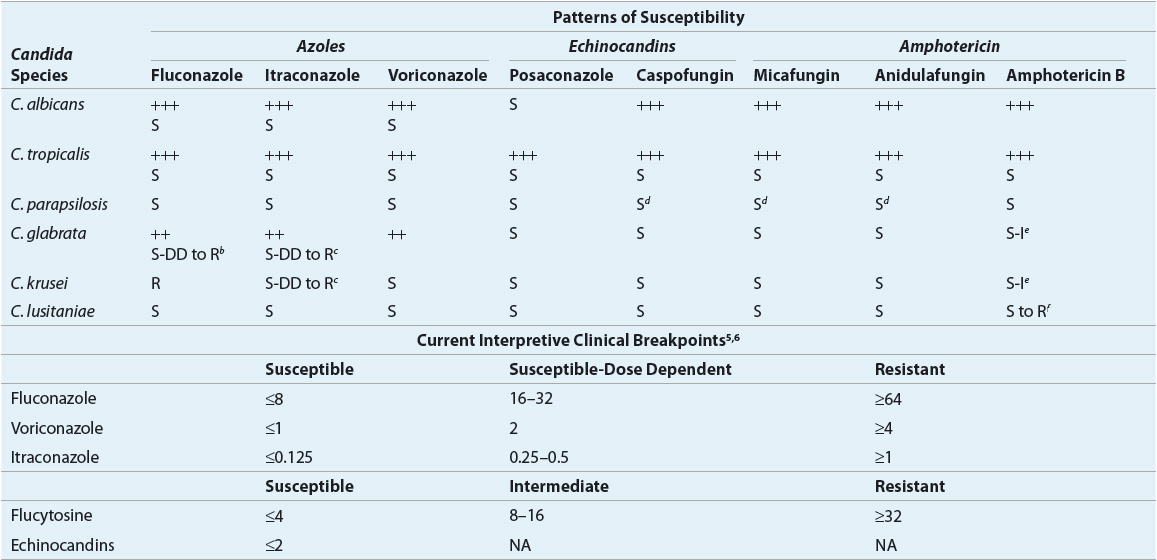
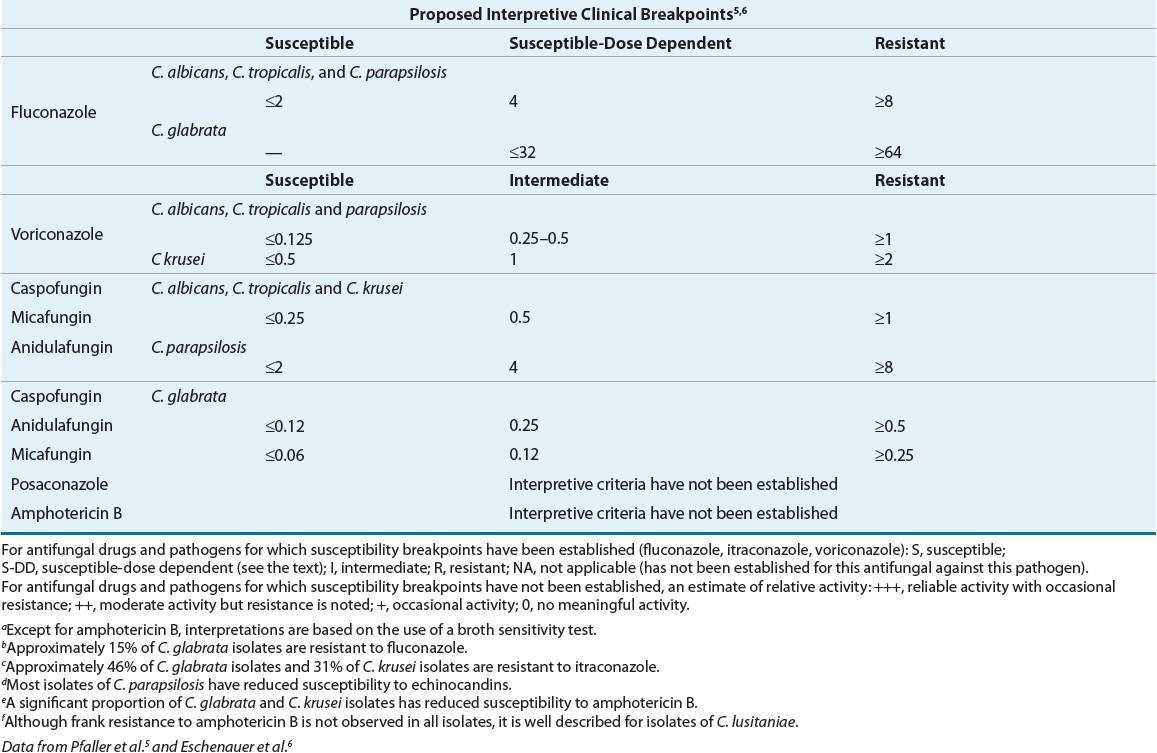
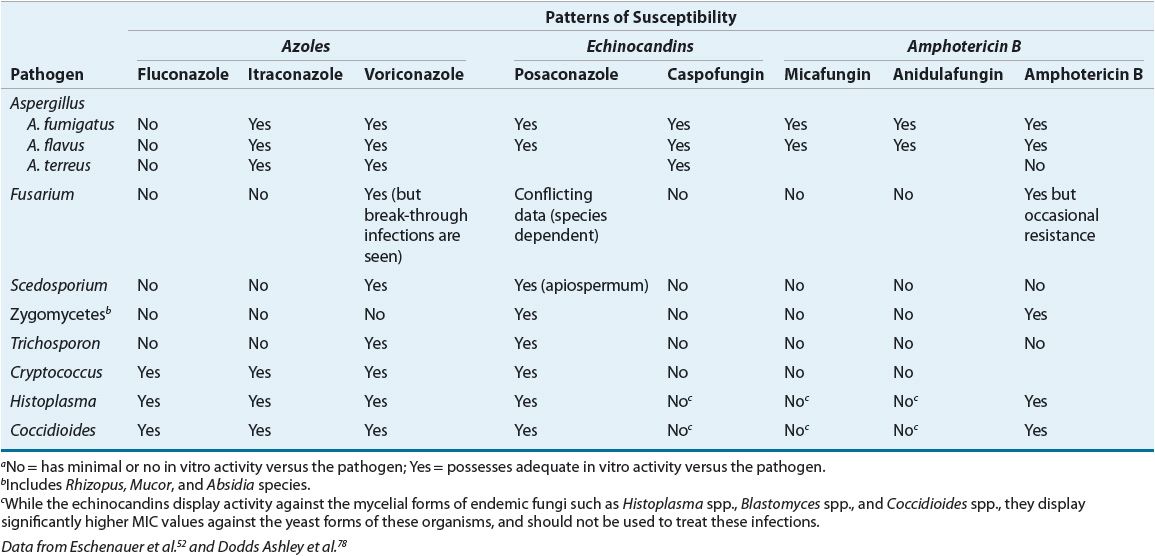
TABLE 99-2 General Patterns of In Vitro Susceptibility of Non-Candida Fungal Pathogensa
Host factors contribute greatly to clinical outcome. A patient may respond clinically to treatment with an antifungal agent despite resistance to that agent in vitro because the patient’s own immune system may eradicate the infection, or the agent may reach the site of infection in high concentrations.13 Thus, in vitro susceptibility does not necessarily equate with in vivo clinical success, and in vitro resistance might not always correlate with treatment failure.
CBPs are based primarily on pharmacokinetic–pharmacodynamic relationships but do take into account other factors, such as differences in dosing regimens, toxicology, resistance mechanisms, intended or approved indications for use, clinical outcome data, and wild type (WT; i.e., the typical strain as it occurs in nature) MIC distributions. CBPs can be used to differentiate strains for which there is a high likelihood of treatment success (organisms which are clinically susceptible, or [S]), from those for which treatment is more likely to fail (clinically resistant [R]). A clinically intermediate (I) or susceptible dose-dependent (SDD) category can be assigned to pathogens for which the level of antimicrobial agent activity is associated with uncertain therapeutic effect, implying that infections due to the isolate may be appropriately treated in body sites where the drugs are physically concentrated or when a high dosage of drug can be used. Although CBPs are designed to guide therapy, they do not distinguish between fungal isolates with or without resistance mechanisms, nor do they always allow for early detection of resistant isolates.
Susceptibility testing occasionally is indicated, for example, in a patient with prolonged fungemia with a presumed susceptible isolate. Because of wide interlaboratory variability in test results, isolates should be tested at specialty laboratories that routinely perform these specialized tests. Susceptibility testing is most helpful in dealing with infections caused by non-albicans species of Candida.5–7
Resistance to Antifungal Agents
It is important to distinguish between clinical resistance and microbial resistance. Clinical resistance refers to failure of an antifungal agent in the treatment of a fungal infection that arises from factors other than microbial resistance, such as failure of the antifungal agent to reach the site of infection or inability of a patient’s immune system to eradicate a fungus whose growth is retarded by an antifungal agent.13
Microbial resistance can refer to primary or secondary resistance, as determined by in vitro susceptibility testing using standardized methodology. Primary or intrinsic resistance refers to resistance recorded prior to drug exposure in vitro or in vivo. Secondary or acquired resistance develops on exposure to an antifungal agent and can be either reversible, owing to transient adaptation, or acquired as a result of one or more genetic alterations. The clinical consequences of antifungal resistance can be observed in treatment failures and in changes in the prevalences of Candida species causing disease.13
The most exhaustive and definitive accounts of antifungal resistance have been described in Candida species, in particular Candida albicans and, to a lesser extent, Candida glabrata, Candida tropicalis, and Candida krusei, as well as in a few C. neoformans isolates.14 There are four different mechanisms that result in azole resistance: (a) mutations or upregulation of ERG11 (an enzyme involved in the ergosterol biosynthesis pathway), (b) expression of multidrug efflux transport pumps that decrease antifungal drug accumulation within the fungal cell, (c) alteration of the structure or concentration of antifungal drug target proteins, and (d) alteration of membrane sterol proteins (Fig. 99–2). It is beyond the scope of this chapter to provide a complete discussion of the biochemical mechanisms of fungal resistance. Interested readers are referred to several excellent reviews concerning this topic.13,14
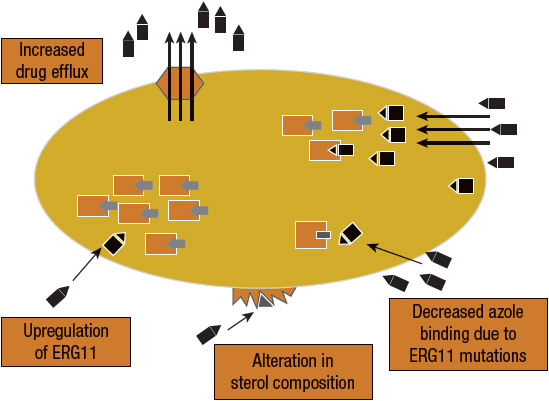
FIGURE 99-2 Mechanisms of azole resistance. Four different mechanisms result in azole resistance: (a) mutations or upregulation of ERG11, the target enzyme of azoles, (b) expression of multidrug efflux transport pumps that decrease antifungal drug accumulation within the fungal cell, (c) alteration of the structure or concentration of antifungal drug target proteins, and (d) alteration of membrane sterol proteins.
Among hospitalized patients, there is increasing evidence for a shift toward isolation of other resistant species, such as C. glabrata and C. krusei, that have moderate or high-level resistance to fluconazole. This phenomenon has been especially common among patients in whom fluconazole has been used extensively.3
The most commonly reported mechanisms of azole resistance among C. albicans isolates include reduced permeability of the fungal cell membrane to azoles, alteration in the target fungal enzymes (cytochrome P450, CYP) resulting in decreased binding of the azole to the target site, and overproduction of the fungal CYP enzymes. Studies also suggest the presence of efflux pumps capable of actively pumping azoles from the target pathogen, thereby conferring multidrug resistance to azole antifungals.13,14 C. glabrata isolates are increasingly resistant to both azole and echinocandin antifungal agents.
Although rare, in vitro intrinsic resistance to amphotericin B is described, primarily in Candida lusitaniae, Candida guilliermondii, and some molds (Fusarium spp. and Pseudallescheria boydii).6 Although the rate of apparent resistance to amphotericin B appears to be quite low, breakthrough bacteremias in patients treated with amphotericin B have been observed. C. glabrata, C. guilliermondii, C. krusei, and C. lusitaniae appear to have a higher propensity than other Candida species to develop resistance to amphotericin B; this point should be kept in mind when treating patients with infections caused by one of these pathogens.6,13 Acquired resistance of Aspergillus species to azoles or echinocandins is relatively uncommon.
Resistant isolates of C. neoformans have been reported to have a mutation in the C8 isomerization step of ergosterol synthesis.6 Current guidelines for the management of cryptococcal infections recommend susceptibility testing only for patients in whom primary treatment has failed, patients with relapse, and for those with recent exposure to antifungals.6 Acquired resistance of Candida species to echinocandins is typically mediated via one of several mechanisms: acquisition of, or intrinsic possession of point mutations in the FKS genes encoding the major subunit of its target enzyme.6
PATHOGENESIS AND EPIDEMIOLOGY
Systemic mycoses caused by primary or pathogenic fungi include histoplasmosis, coccidioidomycosis, cryptococcosis, blastomycosis, paracoccidioidomycosis, and sporotrichosis. Primary pathogens can cause disease in both healthy and immunocompromised individuals, although disease generally is more severe or disseminated in the immunocompromised host. In contrast, mycoses caused by opportunistic fungi such as C. albicans, Aspergillus species, Trichosporon, Torulopsis (Candida) glabrata, Fusarium, Alternaria, and Mucor generally are found only in the immunocompromised host.1
Most fungal infections are acquired as a result of accidental inhalation of airborne conidia. For example, H. capsulatum is found in soil contaminated by bat, chicken, or starling excreta, and C. neoformans is associated with pigeon droppings. Although some fungi, including C. albicans, C. neoformans, and Aspergillus species, are ubiquitous pathogens with worldwide distribution, other fungi have regional distributions associated with specific geographic environments.1
Systemic fungal infections are a major cause of morbidity and mortality in the immunocompromised patient. Fungal infections account for 20% to 30% of fatal infections in patients with acute leukemia, 10% to 15% of fatal infections in patients with lymphoma, and 5% of fatal infections in patients with solid tumors. The frequency of fungal infections among transplant recipients ranges from 0% to 20% for kidney and bone marrow transplant recipients, to 10% to 35% for heart transplant recipients, and 30% to 40% for liver transplant recipients.15,16
Approximately 2% to 4% of all hospitalized patients develop a nosocomial infection. Of these, bacteria comprise the most common etiologic agent.1 Fungi, however, are becoming increasingly significant nosocomial pathogens. Fungi account for 10% of all bloodstream isolates. Candida species (primarily C. albicans) are the fourth most commonly isolated bloodstream isolate and account for 78% of all nosocomial fungal infections.17
Nosocomially acquired fungal infections can arise from either exogenous or endogenous flora. Endogenous flora can include normal commensal organisms of the skin, GI, genitourinary, or respiratory tract. C. albicans is found as a normal commensal of the GI tract in 20% to 30% of humans.
A complex interplay of host and pathogen factors influences the acquisition and development of fungal infections. Intact skin or mucosal surfaces serve as primary barriers to infection. Desiccation, epithelial cell turnover, fatty acid content, and low pH of the skin are believed to be important factors in host resistance. Bacterial flora of the skin and mucous membranes compete with fungi for growth. Alterations in the balance of normal flora caused by the use of antibiotics or alterations in nutritional status can allow the proliferation of fungi such as Candida, increasing the likelihood of systemic invasion and infection.1
Tissue reaction in the presence of fungi varies with fungal species, site of proliferation, and duration of infection. Phagocytosis by neutrophils and macrophages is the earliest mechanism that prevents the establishment of fungi. Consequently, patients with decreased neutrophil counts or decreased neutrophil function are at higher risk of infections, particularly infections caused by Candida and Aspergillus species. Some mycoses are characterized by a low-grade inflammatory response that does not eliminate the fungi. Fungal cells sometimes can persist within macrophages without being killed, perhaps because of resistance to the effects of lysosomal enzymes.1
DIAGNOSIS
![]() The diagnosis of invasive fungal infections generally is accomplished by careful evaluation of clinical symptoms, results of serologic tests, and histopathologic examination and culture of clinical specimens. Skin tests generally are not useful diagnostically because they do not distinguish between active and past infection. They remain useful as screening tools and in epidemiologic studies to determine endemic areas. It is beyond the scope of this chapter to discuss the relative merits of each of the immunologic tests used in the diagnosis of invasive fungal infections. Interested readers, however, are referred to several excellent reviews concerning this topic.18,19
The diagnosis of invasive fungal infections generally is accomplished by careful evaluation of clinical symptoms, results of serologic tests, and histopathologic examination and culture of clinical specimens. Skin tests generally are not useful diagnostically because they do not distinguish between active and past infection. They remain useful as screening tools and in epidemiologic studies to determine endemic areas. It is beyond the scope of this chapter to discuss the relative merits of each of the immunologic tests used in the diagnosis of invasive fungal infections. Interested readers, however, are referred to several excellent reviews concerning this topic.18,19
TREATMENT
Invasive Mycoses
Strategies for the prevention or treatment of invasive mycoses can be classified broadly as prophylaxis, early empirical therapy, empirical therapy, and secondary prophylaxis or suppression.1 In patients undergoing cytotoxic chemotherapy, antifungal therapy is directed primarily at the prevention or treatment of infections caused by Candida and Aspergillus species. Prophylactic therapy with topical, oral, or IV antifungal agents is administered prior to and throughout periods of granulocytopenia (absolute neutrophil count <1,000 cells/L). The potential benefits of prophylactic therapy must be weighed against the potential risks inherent in each regimen, including safety, efficacy, cost, the prevalence of infection, and the potential consequences (e.g., resistance) of widespread use.
Early empirical therapy is the administration of systemic antifungal agents at the onset of fever and neutropenia. Empirical therapy with systemic antifungal agents is administered to granulocytopenic patients with persistent or recurrent fever despite the administration of appropriate antimicrobial therapy.
Secondary prophylaxis (or suppressive therapy) is the administration of systemic antifungal agents (generally prior to and throughout the period of granulocytopenia) to prevent relapse of a documented invasive fungal infection that was treated during a previous episode of granulocytopenia.
Although these treatment classifications also have been applied to the treatment of fungal infections in AIDS, patients with AIDS rarely acquire systemic infections caused by Candida or Aspergillus species, unless they become granulocytopenic because of disease or drugs. The use of antifungal prophylaxis is much less widely studied in this population, although studies suggest that early antifungal prophylaxis with fluconazole or itraconazole decreases the incidence of invasive cryptococcal disease among adult patients who have advanced human immunodeficiency virus (HIV) disease and severe immune suppression (CD4 count <50 cells/mm3 [<50 × 106/L]). However, neither of these interventions showed a clear effect on mortality.9 Suppressive therapy generally is necessary following acute therapy for histoplasmosis, coccidioidomycosis, and cryptococcosis because of the high rates of relapse when antifungal therapy is discontinued.
HISTOPLASMOSIS
In humans, histoplasmosis is caused by inhalation of dust-borne microconidia of the dimorphic fungus H. capsulatum. Although there exist two dimorphic varieties of H. capsulatum, the small-celled (2 to 5 microns) form (var. capsulatum) occurs globally, whereas the large-celled (8 to 15 microns) form (var. duboisii) is confined to the African continent and Madagascar. In tissues stained by conventional techniques, H. capsulatum appears as an oval or round, narrow-pore, budding, unencapsulated yeast.20
Epidemiology
![]() Although histoplasmosis is found worldwide, certain areas of North and Central America are recognized as endemic areas. In the United States, most disease is localized along the Ohio and Mississippi River valleys, where more than 90% of residents may be affected. Precise reasons for this endemic distribution pattern are unknown but are thought to include moderate climate, humidity, and soil characteristics. H. capsulatum is found in nitrogen-enriched soils, particularly those heavily contaminated by avian or bat guano, which accelerates sporulation. Blackbird or pigeon roosts, chicken coops, and sites frequented by bats, such as caves, attics, or old buildings, serve as “microfoci” of infections; once contaminated, soils yield Histoplasma for many years. Although birds are not infected because of their high body temperature, bats (mammals) may be infected and can pass yeast forms in their feces, allowing the spread of H. capsulatum to new habitats. Air currents carry the spores for great distances, exposing individuals who were unaware of contact with the contaminated site.20
Although histoplasmosis is found worldwide, certain areas of North and Central America are recognized as endemic areas. In the United States, most disease is localized along the Ohio and Mississippi River valleys, where more than 90% of residents may be affected. Precise reasons for this endemic distribution pattern are unknown but are thought to include moderate climate, humidity, and soil characteristics. H. capsulatum is found in nitrogen-enriched soils, particularly those heavily contaminated by avian or bat guano, which accelerates sporulation. Blackbird or pigeon roosts, chicken coops, and sites frequented by bats, such as caves, attics, or old buildings, serve as “microfoci” of infections; once contaminated, soils yield Histoplasma for many years. Although birds are not infected because of their high body temperature, bats (mammals) may be infected and can pass yeast forms in their feces, allowing the spread of H. capsulatum to new habitats. Air currents carry the spores for great distances, exposing individuals who were unaware of contact with the contaminated site.20
Pathophysiology
At ambient temperatures, H. capsulatum grows as a mold. The mycelial phase consists of septate branching hyphae with terminal micro- and macroconidia that range in size from 2 to 14 microns in diameter. When soil is disturbed, these conidia become aerosolized and reach the bronchioles or alveoli.20
Animal studies demonstrate that within 2 to 3 days after reaching lung tissue, the conidia germinate, releasing yeast forms that begin multiplying by binary fission. During the next 9 to 15 days, organisms are ingested but not destroyed by large numbers of macrophages that are recruited to the infected site, resulting in small infiltrates. Infected macrophages migrate to the mediastinal lymph nodes and other sites within the mononuclear phagocyte system, particularly the spleen and liver. At this time, the onset of specific T-cell immunity in the nonimmune host activates the macrophages, rendering them capable of fungicidal activity. Tissue granulomas form, many of which develop central caseation and necrosis over the next 2 to 4 months. Over a period of several years, these foci become encapsulated and calcified, often with viable yeast trapped within the necrotic tissue.20,21
Cellular immunity, as measured by histoplasmin skin-test reactivity, wanes in the absence of occasional reexposure. Although exposure to heavy inocula can overcome these immune mechanisms, resulting in severe disease, reinfection occurs frequently in endemic areas. In the immune individual, the reactions of acquired immunity begin 24 to 48 hours after the appearance of yeast forms, resulting in milder forms of illness and little proliferation of organisms. Although viable organisms can be found within granulomas years after initial infection, the organisms appear to have little ability to proliferate within the fibrous capsules, except in immunocompromised patients.20,21
Clinical Presentation
The outcome of infection with H. capsulatum depends on a complex interplay of host, pathogen, and environmental factors.10,20,21 Host factors include the degree of immunosuppression and the presence of immunity (from prior infection). Environmental factors include inoculum size, exposure within an enclosed area, and duration of exposure. Hematogenous dissemination from the lungs to other tissues probably occurs in all infected individuals during the first 2 weeks of infection before specific immunity has developed but is nonprogressive in most cases, which leads to the development of calcified granulomas of the liver and/or spleen. Progressive pulmonary infection is common in patients with underlying centrilobular emphysema.
Acute and chronic manifestations of histoplasmosis appear to result from unusual inflammatory or fibrotic responses to the pathogen, including pericarditis and rheumatologic syndromes during the first year after exposure, with chronic mediastinal inflammation or fibrosis, broncholithiasis, and enlarging parenchymal granulomas later in the course of disease.
Acute Pulmonary Histoplasmosis
In the vast majority of patients, low-inoculum exposure to H. capsulatum results in mild or asymptomatic pulmonary histoplasmosis. The course of disease generally is benign, and symptoms usually abate within a few weeks of onset. Patients exposed to a higher inoculum during an acute primary infection or reinfection can experience an acute, self-limited illness with flu-like pulmonary symptoms, including fever, chills, headache, myalgia, and a nonproductive cough. Patients with diffuse pulmonary histoplasmosis can have diffuse radiographic involvement, become hypoxic, and require ventilatory support. A low percentage of patients present with arthritis, erythema nodosum, pericarditis, or mediastinal granuloma.
Chronic Pulmonary Histoplasmosis
Chronic pulmonary histoplasmosis generally presents as an opportunistic infection imposed on a preexisting structural abnormality, such as lesions resulting from emphysema. Patients demonstrate chronic pulmonary symptoms and apical lung lesions that progress with inflammation, calcified granulomas, and fibrosis. Patients with early, noncavitary disease often recover without treatment. Progression of disease over a period of years, seen in 25% to 30% of patients, is associated with cavitation, bronchopleural fistulas, extension to the other lung, pulmonary insufficiency, and often death.
Disseminated Histoplasmosis
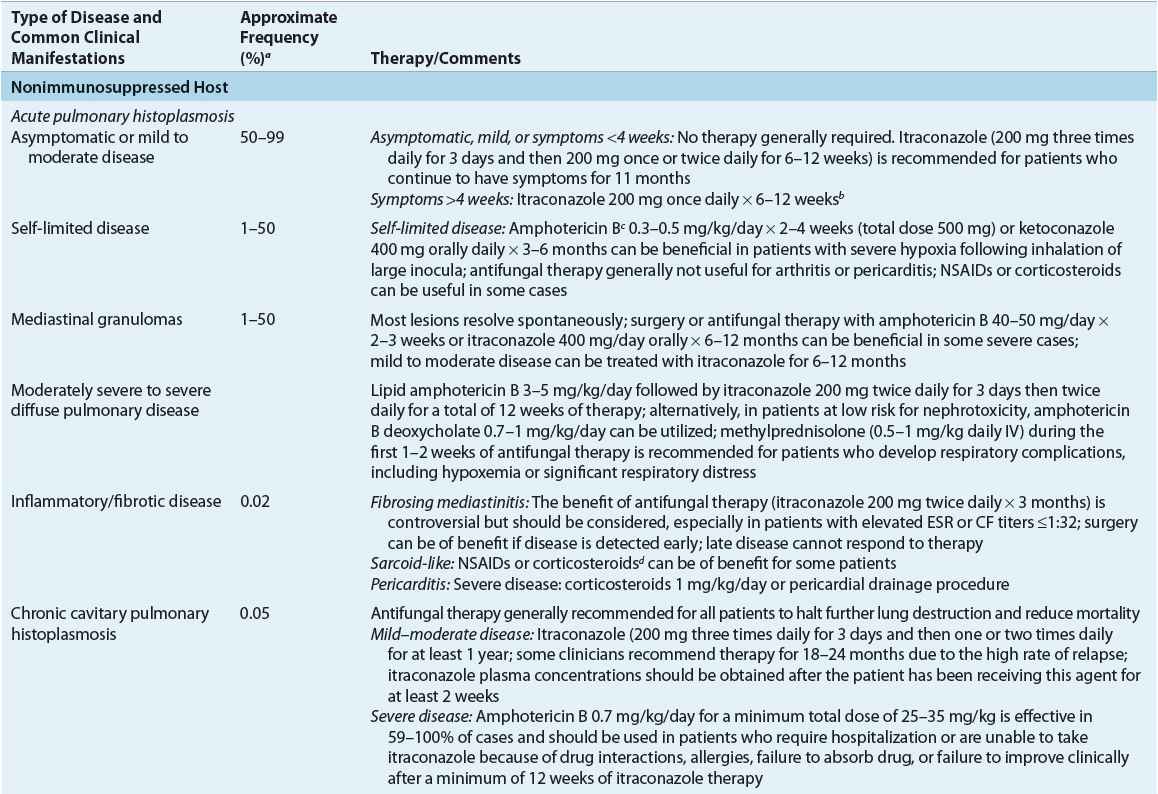
In patients exposed to a large inoculum and in immunocompromised hosts, successful containment of the organism within macrophages may not occur, resulting in a progressive illness characterized by yeast-filled phagocytic cells and an inability to produce granulomas. This disease, termed disseminated histoplasmosis, is characterized by persistent parasitization of macrophages. The clinical severity of the diverse forms of disseminated histoplasmosis (Table 99–3) generally parallels the degree of macrophage parasitization observed.
TABLE 99-3 Clinical Manifestations and Therapy of Histoplasmosis
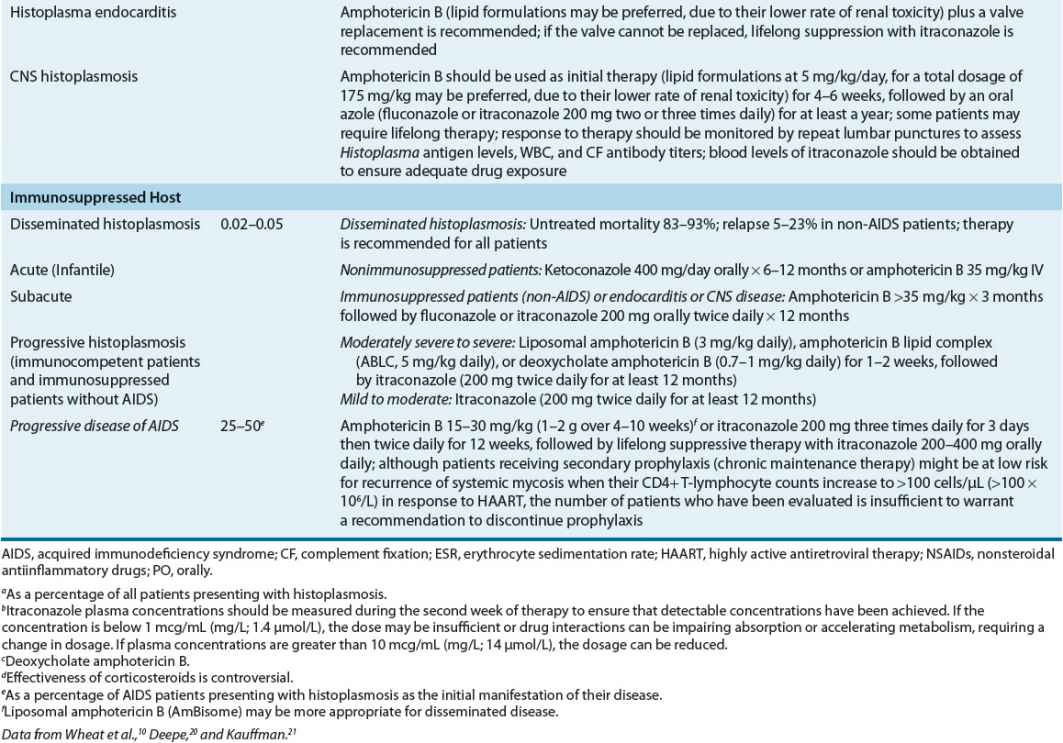
Acute (infantile) disseminated histoplasmosis is characterized by massive involvement of the mononuclear phagocyte system by yeast-engorged macrophages. Classically, this severe type of infection is seen in infants and young children and (rarely) in adults with Hodgkin’s disease or other lymphoproliferative disorders. In infants or children, acute disseminated histoplasmosis is characterized by unrelenting fever, anemia, leukopenia or thrombocytopenia, enlargement of the liver, spleen, and visceral lymph nodes, and GI symptoms, particularly nausea, vomiting, and diarrhea. The chest roentgenogram often demonstrates remnants of the initiating acute pulmonary lesion. Untreated disease is uniformly fatal in 1 to 2 months. A less severe “subacute” form of the disease, which occurs in both infants and immunocompetent adults, is characterized by focal destructive lesions in various organs, weight loss, weakness, fever, and malaise. Untreated disease generally is fatal in approximately 10 months.
Most adults with disseminated histoplasmosis demonstrate a mild, chronic form of the disease. Untreated patients often are ill for 10 to 20 years, demonstrating long asymptomatic periods interrupted by relapses of clinical illness characterized primarily by weight loss, weakness, and fatigue. Chronic disseminated histoplasmosis can be seen in patients with lymphoreticular neoplasms (Hodgkin’s disease) and patients undergoing immunosuppressant chemotherapy for organ transplantation or for rheumatic diseases. Although CNS involvement occurs in 10% to 20% of patients with severe underlying immunosuppressive conditions, focal organ involvement is uncommon. The disease is characterized by the development of focal granulomatous lesions, often with bone marrow involvement resulting in thrombocytopenia, anemia, and leukemia. Fever, hepatosplenomegaly, and GI ulceration are common.
Histoplasmosis in HIV-Infected Patients
Adult patients with AIDS demonstrate an acute form of disseminated disease that resembles the syndrome seen in infants and children. Progressive disseminated histoplasmosis (PDH), which is defined as a clinical illness that does not improve after at least 3 weeks of observation and that is associated with physical or radiographic findings and/or laboratory evidence of involvement of extrapulmonary tissues, can occur as the direct result of initial infection or because of the reactivation of dormant foci. In endemic areas, 50% of AIDS patients demonstrate PDH as the first manifestation of their disease. PDH is characterized by fever (75% of patients), weight loss, chills, night sweats, enlargement of the spleen, liver, or lymph nodes, and anemia. Pulmonary symptoms occur in only one third of patients and do not always correlate with the presence of infiltrates on chest roentgenogram. A clinical syndrome resembling septicemia is seen in approximately 25% to 50% of patients.21
Diagnosis
Detection of single, yeastlike cells 2 to 5 microns in diameter with narrow-based budding by direct examination or by histologic study of blood smears or tissues should raise strong suspicion of infection with H. capsulatum because colonization does not occur as with Aspergillus or Candida infection. Identification of mycelial isolates from clinical cultures can be made by conversion of the mycelium to the yeast form (requires 3 to 6 weeks) or through a rapid (2 hours) and 100% sensitive chemiluminescent DNA probe that recognizes ribosomal DNA. In patients with suspected disseminated or chronic cavitary histoplasmosis, 2 to 3 blood, sputum, and bone marrow cultures and stains should be obtained using the lysis centrifugation (Isolator tube) technique, and the cultures should be held for 14 to 21 days for optimal yield of H. capsulatum. In patients with acute self-limited histoplasmosis, extensive testing to verify the diagnosis may not be necessary.
In most patients, serologic evidence remains the primary method in the diagnosis of histoplasmosis. Results obtained from commercially available complement fixation (CF), immunodiffusion (ID), and latex agglutination (LA) antibody tests are used alone or in combination. In general, the use of histoplasmin skin tests is of little value except in epidemiologic studies because histoplasmin reactivity waxes in the absence of occasional reexposure. In addition, histoplasmin skin testing can result in a false increase in the CF titer for mycelial antigen (CF-M) to H. capsulatum. A fourfold rise in the CF titer is usually indicative of recent infection, although some patients with severe disease or profound immunosuppression can demonstrate a weaker antibody response. CF titers remain positive for many years, since CF antibodies persist after infection. Because the ID test is more specific but less sensitive than CF, it should be used to assess the importance of weakly reactive results obtained by CF rather than as a screening procedure.
In the AIDS patient with PDH, the diagnosis is best established by bone marrow biopsy and culture, which yield positive cultures in more than 90% of patients, although blood cultures and histopathologic examination and culture of pulmonary tissue, sputum, skin, and lymph nodes also can be helpful. Detection of H. capsulatum polysaccharide antigen (HPA) in urine, blood, or cerebrospinal fluid (CSF) by enzyme-linked immunosorbent assay (ELISA) or by modified radioimmunoassay (RIA) offer promising new techniques for the rapid diagnosis of histoplasmosis. The HPA (by RIA) levels also have been used successfully to monitor the course of therapy and to detect relapses in patients with AIDS, and the clearance of antigen from serum and urine correlates with clinical efficacy during maintenance therapy with itraconazole.21
TREATMENT
Non–HIV-Infected Patient
![]() Table 99–3 summarizes the recommended therapy for the treatment of histoplasmosis. In general, asymptomatic or mildly ill patients and patients with sarcoid-like disease do not benefit from antifungal therapy. In the vast majority of patients, low-inoculum exposure to H. capsulatum results in mild or asymptomatic pulmonary histoplasmosis. The course of disease generally is benign, and symptoms usually abate within a few weeks of onset. Therapy can be helpful in symptomatic patients whose conditions have not improved during the first month of infection. Fever persisting more than 3 weeks can indicate that the patient is developing progressive disseminated disease, which can be aborted by antifungal therapy. Whether antifungal therapy hastens recovery or prevents complications is unknown because it has never been studied in prospective trials.
Table 99–3 summarizes the recommended therapy for the treatment of histoplasmosis. In general, asymptomatic or mildly ill patients and patients with sarcoid-like disease do not benefit from antifungal therapy. In the vast majority of patients, low-inoculum exposure to H. capsulatum results in mild or asymptomatic pulmonary histoplasmosis. The course of disease generally is benign, and symptoms usually abate within a few weeks of onset. Therapy can be helpful in symptomatic patients whose conditions have not improved during the first month of infection. Fever persisting more than 3 weeks can indicate that the patient is developing progressive disseminated disease, which can be aborted by antifungal therapy. Whether antifungal therapy hastens recovery or prevents complications is unknown because it has never been studied in prospective trials.
Fluconazole remains a second-line agent for the treatment of histoplasmosis. Clinical data regarding the use of newer azoles such as voriconazole and posaconazole are limited. While both have activity against Histoplasma, posaconazole appears to be more active than itraconazole in the immune compromised and nonimmune compromised mouse model of infection, while voriconazole has not been tested in animal models. Both agents have been used successfully in a few patients. Of note, the echinocandins have no activity against Histoplasma.
Patients with mild, self-limited disease, chronic disseminated disease, or chronic pulmonary histoplasmosis who have no underlying immunosuppression usually can be treated with either oral itraconazole or IV amphotericin B. The goals of therapy are resolution of clinical abnormalities, prevention of relapse, and eradication of infection whenever possible, although chronic suppression of infection can be adequate in immunosuppressed patients, including those with HIV disease.10,21
HIV-Infected Patient
In AIDS patients, intensive 12-week primary antifungal therapy (induction and consolidation therapy) is followed by lifelong suppressive (maintenance) therapy with itraconazole. Amphotericin B dosages of 50 mg/day (up to 1 mg/kg per day) should be administered IV to a cumulative dose of 15 to 35 mg/kg (1 to 2 g) in patients who require hospitalization. Amphotericin B can be replaced with itraconazole 200 mg orally twice daily when the patient no longer requires hospitalization or IV therapy to complete a 12-week total course of induction therapy. In patients who do not require hospitalization, itraconazole therapy for 12 weeks can be used.
Fluconazole 800 mg/day orally as induction, followed by 400 mg/day, was effective in 88% of patients, but relapses occurred in approximately one third of patients, and in vitro resistance developed in approximately 50% of patients who relapsed.
In regions experiencing high rates of histoplasmosis (>5 cases/100 patient-years), itraconazole 200 mg/day is recommended as prophylactic therapy in HIV-infected patients. Fluconazole is not an acceptable alternative because of its inferior activity against H. capsulatum and its lower efficacy for the treatment of histoplasmosis.10
Although patients receiving secondary prophylaxis (chronic maintenance therapy) might be at low risk for recurrence of systemic mycosis when their CD4+ T lymphocyte counts increase to >100 cells/μL (>100 × 106/L) in response to highly active antiretroviral therapy (HAART), the number of patients who have been evaluated is insufficient to warrant a recommendation to discontinue prophylaxis.
Evaluation of Therapeutic Outcomes
Response to therapy should be measured by resolution of radiologic, serologic, and microbiologic parameters and by improvement in signs and symptoms of infection. Although investigators are limited by the lack of standardized criteria to quantify the extent of infection, degree of immunosuppression, or treatment response, response rates (based on resolution or improvement in presenting signs and symptoms) of greater than 80% have been reported in case series in AIDS patients receiving varied dosages of amphotericin B. Rapid responses are reported, with the resolution of symptoms in 25% and 75% of patients by days 3 and 7 of therapy, respectively.
After the initial course of therapy for histoplasmosis is complete, lifelong suppressive therapy with oral azoles or amphotericin B (1 to 1.5 mg/kg weekly or biweekly) is recommended because of the frequent recurrence of infection. Relapse rates in AIDS patients not receiving maintenance therapy range from 50% to 90%.10
Antigen testing can be useful for monitoring therapy in patients with disseminated histoplasmosis. Antigen concentrations decrease with therapy and increase with relapse.
BLASTOMYCOSIS
North American blastomycosis is a systemic fungal infection caused by Blastomyces dermatitidis, a dimorphic fungus that infects primarily the lungs. Patients, however, can present with a variety of pulmonary and extrapulmonary clinical manifestations. Pulmonary disease can be acute or chronic and can mimic infection with tuberculosis, pyogenic bacteria, other fungi, or malignancy. Blastomycosis can disseminate to virtually every other body organ, and approximately 40% of patients with blastomycosis present with skin, bone and joint, or genitourinary tract involvement without any evidence of pulmonary disease.8,22
Pulmonary infection probably occurs by inhalation of conidia, which convert to the yeast form in the lung. A vigorous inflammatory response ensues, with neutrophilic recruitment to the lungs followed by the development of cell-mediated immunity and the formation of noncaseating granulomas.
Epidemiology
Blastomycosis was renamed North American blastomycosis in 1942, when Conant and Howell named a similar fungus endemic to South America, Blastomyces braziliensis, and the disease it caused South American blastomycosis. Although the disease is now recognized to be endemic to the southeastern and south central states of the United States (especially those bordering on the Mississippi and Ohio River basins) and the midwestern states and Canadian provinces bordering the Great Lakes, numerous cases of North American blastomycosis have been diagnosed in Africa, northern parts of South America, India, and Europe. Endemic areas have been defined primarily by analysis of sporadic cases and epidemics or clusters of disease because the lack of a dependable skin or laboratory test makes wide-scale epidemiologic testing to determine the incidence of infection unfeasible at present.8,19,22 Although initial review of sporadic cases suggested that males with outdoor occupations that exposed them to soil were at greatest risk for blastomycosis, there is no sex, age, or occupational predilection for blastomycosis.8,19,22
Although B. dermatitidis generally is considered to be a soil inhabitant, attempts to isolate the organism in nature frequently have been unsuccessful. B. dermatitidis has been isolated from soil containing decayed vegetation, decomposed wood, and pigeon manure, frequently in association with warm, moist soil of wooded areas that is rich in organic debris.8,19,22
Pathophysiology and Clinical Presentation
Colonization does not occur with Blastomyces.8,19,22 Acute pulmonary blastomycosis generally is an asymptomatic or self-limited disease characterized by fever, shaking chills, and productive, purulent cough, with or without hemoptysis, in immunocompetent individuals. The clinical presentation can be difficult to differentiate from other respiratory infections, including bacterial pneumonia, on the basis of clinical symptoms alone.
Sporadic (nonepidemic) pulmonary blastomycosis can present as a more chronic or subacute disease, with low-grade fever, night sweats, weight loss, and productive cough that resembles tuberculosis rather than bacterial pneumonia. Chronic pulmonary blastomycosis is characterized by fever, malaise, weight loss, night sweats, chest pain, and productive cough. Patients often are thought to have tuberculosis and frequently have evidence of disseminated disease that can appear 1 to 3 years after the primary pneumonia has resolved. Reactivation of disease can occur in the lungs or as the focus of new infection in other organs.
In approximately 40% of patients, dissemination is not accompanied by reactivation of pulmonary disease. The most common sites for disseminated disease include the skin and bony skeleton, although less commonly the prostate, oropharyngeal mucosa, and abdominal viscera are involved. CNS disease, while exceedingly uncommon, is associated with the highest mortality rate.
Laboratory and Diagnostic Tests
The simplest and most successful method of diagnosing blastomycosis is by direct microscopic visualization of the large, multinucleated yeast with single, broad-based buds in sputum or other respiratory specimens following digestion of cells and debris with 10% potassium hydroxide.8,19 Histopathologic examination of tissue biopsies and culture of secretions also should be used to identify B. dermatitidis, although it can require up to 30 days to isolate and identify a small inoculum.
No reliable skin test exists to determine the incidence and prevalence of disease in endemic populations, and reliable serologic diagnosis of blastomycosis has long been hampered by the lack of specific and standardized reagents. Serologic response does not always correlate with clinical improvement, although some investigators have noted that a decline in the number of precipitins or CF titers can offer evidence of a favorable prognosis in patients with established disease.
Acute pulmonary blastomycosis generally is an asymptomatic or self-limited disease characterized by fever, shaking chills, and productive, purulent cough, with or without hemoptysis, in immunocompetent individuals. The clinical presentation can be difficult to differentiate from other respiratory infections, including bacterial pneumonia, on the basis of clinical symptoms alone. Sporadic (nonepidemic) cases of pulmonary blastomycosis can present as a more chronic or subacute disease with low-grade fever, night sweats, weight loss, and productive cough that resembles tuberculosis rather than bacterial pneumonia.
TREATMENT
Non–HIV-Infected Patient
![]() In the immunocompetent host, acute pulmonary blastomycosis can be mild and self-limited and may not require treatment. However, consideration should be given to treating all infected individuals to prevent extrapulmonary dissemination. All individuals with moderate to severe pneumonia, disseminated infection, or those who are immunocompromised require antifungal therapy.
In the immunocompetent host, acute pulmonary blastomycosis can be mild and self-limited and may not require treatment. However, consideration should be given to treating all infected individuals to prevent extrapulmonary dissemination. All individuals with moderate to severe pneumonia, disseminated infection, or those who are immunocompromised require antifungal therapy.
In patients with mild to moderate pulmonary blastomycosis, itraconazole is effective; however, in patients with moderately severe to severe pulmonary disease, the clinical presentation of the patient, the immune competence of the patient, and the toxicity of the antifungal agents are the main determinants of the choice of antifungal therapy. All immunocompromised patients and patients with progressive pulmonary disease or with extrapulmonary disease should be treated (Table 99–4). In the case of disease limited to the lungs, cure might have occurred without treatment before the diagnosis is made. Regardless of whether or not the patient receives treatment, however, he or she must be followed carefully for many years for evidence of reactivation or progressive disease.8,19,22
TABLE 99-4 Therapy of Blastomycosis
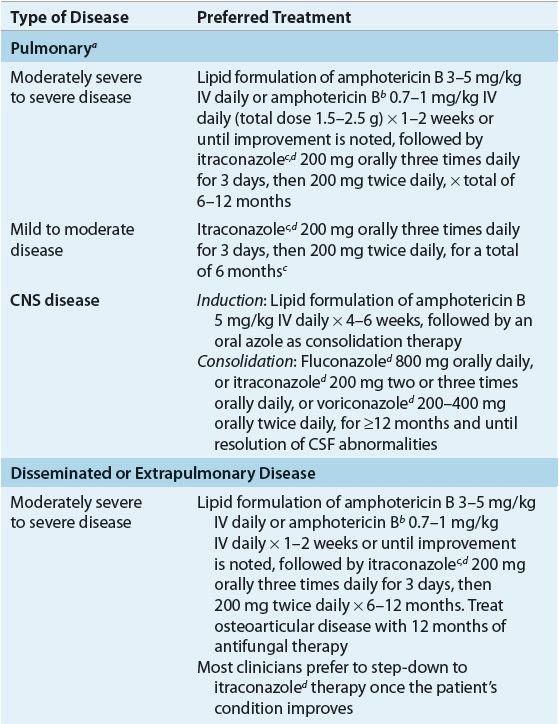

Some authors recommend azole therapy for the treatment of self-limited pulmonary disease, with the hope of preventing late extrapulmonary disease; however, data supporting the efficacy of these regimens are lacking.8,22 Itraconazole 200 to 400 mg/day demonstrated 90% efficacy as a first-line agent in the treatment of non–life-threatening non-CNS blastomycosis, and for compliant patients who completed at least 2 months of therapy, a success rate of 95% was noted. No therapeutic advantage was noted with the higher (400 mg) dosage as compared with patients treated with 200 mg.
All patients with disseminated blastomycosis, as well as those with extrapulmonary disease, require therapy. Due to its adverse effects, variable oral absorption, and lack of CNS penetration, ketoconazole is now reserved as an alternative therapy for mild to moderate pulmonary and non-CNS disease. However, older studies demonstrate that ketoconazole 400 mg/day orally for 6 months cures more than 80% of patients with chronic pulmonary and nonmeningeal disseminated blastomycosis. Amphotericin B is more efficacious but more toxic and therefore is reserved for noncompliant patients and patients with overwhelming or life-threatening disease, CNS infection, and treatment failures.8,22 Lipid preparations of amphotericin B have largely replaced conventional amphotericin B for treatment of blastomycosis, despite their higher cost, due to their decreased renal toxicity. Surgery has only a limited role in the treatment of blastomycosis.
HIV-Infected Patient
For unclear reasons, blastomycosis is an uncommon opportunistic disease among immunocompromised individuals, including AIDS patients; however, blastomycosis can occur as a late (CD4 lymphocytes <200 cells/mm3 [<200 × 106/L]) and frequently fatal complication of HIV infection. In this population, overwhelming disseminated disease with frequent involvement of the CNS is common.8,22 Following induction therapy with amphotericin B (total cumulative dose of 1 g), HIV-infected patients should receive chronic suppressive therapy with an oral azole antifungal.8,22
COCCIDIOIDOMYCOSIS
Epidemiology
Coccidioidomycosis is caused by infection with Coccidioides immitis, a dimorphic fungus found in the southwestern and western United States, as well as in parts of Mexico and South America. In North America, the endemic regions encompass the semiarid areas of the southwestern United States from California to Texas known as the Lower Sonoran Zone, where there is scant annual rainfall, hot summers, and sandy, alkaline soil. C. immitis grows in the soil as a mold, and mycelia proliferate during the rainy season. During the dry season, resistant arthroconidia form and become airborne when the soil is disturbed.
Although generally considered to be a regional disease, coccidioidomycosis has increased in importance in recent years because of the increased tourism and population in endemic areas, the increased use of immunosuppressive therapy in transplantation and oncology, and the AIDS epidemic. Although there is no racial, hormonal, or immunologic predisposition for acquiring primary disease, these factors affect the risk of subsequent dissemination of disease (Table 99–5).11
TABLE 99-5 Factors for Severe, Disseminated Infection with Coccidioidomycosis

Pathophysiology
When individuals come in contact with contaminated soil during ranching, dust storms, or proximity to construction sites or archaeologic excavations, arthroconidia are inhaled into the respiratory tree, where they transform into spherules, which reproduce by cleavage of the cytoplasm to produce endospores. The endospores are released when the spherules reach maturity. Similar to histoplasmosis, an acute inflammatory response in the tissue leads to infiltration of mononuclear cells, ultimately resulting in granuloma formation.11
Clinical Presentation of Coccidioidomycosis
Coccidioidomycosis encompasses a spectrum of illnesses ranging from primary uncomplicated respiratory tract infection that resolves spontaneously to progressive pulmonary or disseminated infection.11,19,23 Initial or primary infection with C. immitis almost always involves the lungs. Although approximately one third of the population in endemic areas is infected, the average incidence of symptomatic disease is only approximately 0.43%.
Signs and Symptoms
Primary Coccidioidomycosis (“Valley Fever”) Approximately 60% of infected patients have an asymptomatic, self-limited infection without clinical or radiological manifestations. The remaining 40% of patients exhibit nonspecific symptoms that are often indistinguishable from ordinary upper respiratory infections, including fever, cough, headache, sore throat, myalgias, and fatigue that occur 1 to 3 weeks after exposure to the pathogen. More commonly, a diffuse, mild erythroderma or maculopapular rash is observed. Patients can have pleuritic chest pain and peripheral eosinophilia.
A fine, diffuse rash can appear during the first few days of the illness. Primary pneumonia can be the first manifestation of disease, characterized by a productive cough that can be blood-streaked, as well as single or multiple soft or dense homogeneous hilar or basal infiltrates on chest roentgenogram. Chronic, persistent pneumonia or persistent pulmonary coccidioidomycosis (primary disease lasting more than 6 weeks) is complicated by hemoptysis, pulmonary scarring, and the formation of cavities or bronchopleural fistulas.
Necrosis of pulmonary tissue with drainage and cavity formation occurs commonly. Most parenchymal cavities close spontaneously or form dense nodular scar tissue that can become superinfected with bacteria or spherules of C. immitis. These patients often have persistent cough, fevers, and weight loss.
Disseminated disease occurs in less than 1% of infected patients. The most common sites for dissemination are the skin, lymph nodes, bone, and meninges, although the spleen, liver, kidney, and adrenal gland also can be involved. Occasionally, miliary coccidioidomycosis occurs, with rapid, widespread dissemination, often in concert with positive blood cultures for C. immitis. Patients with AIDS frequently present with miliary disease. Coccidioidomycosis in AIDS patients appears to be caused by reactivation of disease in most patients. Dissemination also is more likely if infection occurs during pregnancy, especially during the third trimester or in the immediate postpartum period.23
CNS infection occurs in approximately 16% of patients with disseminated coccidioidomycosis. Patients can present with meningeal disease without previous symptoms of primary pulmonary infection, although disease usually occurs within 6 months of the primary infection. The signs and symptoms are often subtle and nonspecific, including headache, weakness, changes in mental status (lethargy and confusion), neck stiffness, low-grade fever, weight loss, and occasionally, hydrocephalus. Space-occupying lesions are rare, and the main areas of involvement are the basilar meninges.
Diagnosis
The diagnoses of coccidioidomycosis generally utilizes identification or recovery of Coccidioides spp. from clinical specimens and detection of specific anticoccidioidal antibodies in serum or other body fluids.19
TREATMENT
General Guidelines
![]() Therapy for coccidioidomycosis is difficult, and the results are unpredictable. Guidelines11 are available for treatment of this disease; however, optimal treatment for many forms of this disease still generates debate. The efficacy of antifungal therapy for coccidioidomycosis often is less certain than that for other fungal etiologies, such as blastomycosis, histoplasmosis, or cryptococcus, even when in vitro susceptibilities and the sites of infections are similar. The refractoriness of coccidioidomycosis can relate to the ability of C. immitis spherules to release hundreds of endospores, maximally challenging host defenses.11,23 Fortunately, only approximately 5% of infected patients require therapy.23
Therapy for coccidioidomycosis is difficult, and the results are unpredictable. Guidelines11 are available for treatment of this disease; however, optimal treatment for many forms of this disease still generates debate. The efficacy of antifungal therapy for coccidioidomycosis often is less certain than that for other fungal etiologies, such as blastomycosis, histoplasmosis, or cryptococcus, even when in vitro susceptibilities and the sites of infections are similar. The refractoriness of coccidioidomycosis can relate to the ability of C. immitis spherules to release hundreds of endospores, maximally challenging host defenses.11,23 Fortunately, only approximately 5% of infected patients require therapy.23
Goals of Therapy
Desired outcomes of treatment are resolution of signs and symptoms of infection, reduction of serum concentrations of anticoccidioidal antibodies, and return of function of involved organs. It would also be desirable to prevent relapse of illness on discontinuation of therapy, although current therapy is often unable to achieve this goal.
Specific Agents Used for the Treatment of Coccidioidomycosis
Azole antifungals, primarily fluconazole and itraconazole, have replaced amphotericin B as initial therapy for most chronic pulmonary or disseminated infections. Amphotericin B is now usually reserved for patients with respiratory failure because of infection with Coccidioides species, those with rapidly progressive coccidioidal infections, or women during pregnancy. Therapy often ranges from many months to years in duration, and in some patients, lifelong suppressive therapy is needed to prevent relapses. Specific antifungals (and their usual dosages) for the treatment of coccidioidomycosis include IV amphotericin B (0.5 to 1.5 mg/kg per day), ketoconazole (400 mg/day orally), IV or oral fluconazole (usually 400 to 800 mg/day, although dosages as high as 1,200 mg/day have been used without complications), and itraconazole (200 to 300 mg orally twice daily or three times daily, as either capsules or solution).11,23 If itraconazole is used, measurement of serum concentrations can be helpful to ascertain whether oral bioavailability is adequate.
Amphotericin B generally is preferred as initial therapy in patients with rapidly progressive disease, whereas azoles generally are preferred in patients with subacute or chronic presentations. The lipid formulations of amphotericin B have not been studied extensively in coccidioidal infection but can offer a means of giving more drug with less toxicity. Fluconazole probably is the most frequently used medicine given its tolerability, although high relapse rates have been reported in some studies. Relapse rates with itraconazole therapy can be lower than those with fluconazole.11,23
The usefulness of newly available antifungal agents of possible benefit for the treatment of refractory coccidioidal infections has not been adequately assessed and they are not yet FDA approved for use in this population. Case reports have suggested that voriconazole can be effective in selected patients. Caspofungin has been effective in treating experimental murine coccidioidomycosis, but in vitro susceptibility of isolates varies widely, and there is only one report regarding its value. Posaconazole was shown to be an effective treatment in a small clinical trial and in patients with refractory infections. Its efficacy relative to other triazole antifungals is unknown.
Clinical Controversy…
Combination therapy with members of different classes of antifungal agents has not been evaluated in patients, and there is a hypothetical risk of antagonism. However, some clinicians feel that outcome in severe cases is improved when amphotericin B is combined with an azole antifungal. If the patient improves, the dosage of amphotericin B can be slowly decreased while the dosage of azole is maintained.11,23
Primary Respiratory Infection
Although most patients with symptomatic primary pulmonary disease recover without therapy, management should include followup visits for 1 to 2 years to document resolution of disease or to identify as early as possible evidence of pulmonary or extrapulmonary complications.
Patients with a large inoculum, severe infection, or concurrent risk factors (e.g., HIV infection, organ transplant, pregnancy, or high doses of corticosteroids) probably should be treated, particularly those with high CF titers, in whom incipient or occult dissemination is likely. Because some racial or ethnic populations have a higher risk of dissemination, some clinicians advocate their inclusion in the high-risk group. Common indicators used to judge the severity of infection include weight loss (>10%), intense night sweats persisting more than 3 weeks, infiltrates involving more than one half of one lung or portions of both lungs, prominent or persistent hilar adenopathy, CF antibody titers of greater than 1:16, failure to develop dermal sensitivity to coccidial antigens, inability to work, or symptoms that persist for more than 2 months.11,23
Commonly prescribed therapies include currently available oral azole antifungals at their recommended doses for courses of therapy ranging from 3 to 6 months.11,23 In patients with diffuse pneumonia with bilateral reticulonodular or miliary infiltrates, therapy usually is initiated with amphotericin B; several weeks of therapy generally are required to produce clear evidence of improvement. Consolidation therapy with oral azoles can be considered at that time. The total duration of therapy should be at least 1 year, and in patients with underlying immunodeficiency, oral azole therapy should be continued as secondary prophylaxis. Although HIV-infected patients receiving secondary prophylaxis might be at low risk for recurrence of systemic mycosis when their CD4+ T-lymphocyte counts increase to >100 cells/μL (>100 × 106/L) in response to HAART, the number of patients who have been evaluated is insufficient to warrant a recommendation to discontinue prophylaxis.
Infections of the Pulmonary Cavity
Many pulmonary infections that are caused by C. immitis are benign in their course and do not require intervention. In the absence of controlled clinical trials, evidence of the benefit of antifungal therapy is lacking, and asymptomatic infections generally are left untreated. Symptomatic patients can benefit from oral azole therapy, although recurrence of symptoms can be seen in some patients once therapy is discontinued. Surgical resection of localized cavities provides resolution of the problem in patients in whom the risks of surgery are not too high.11,23
Extrapulmonary (Disseminated) Disease
Nonmeningeal Disease
Almost all patients with disease located outside the lungs should receive antifungal therapy; therapy usually is initiated with 400 mg/day of an oral azole. Amphotericin B is an alternative therapy and can be necessary in patients with worsening lesions or with disease in particularly critical locations such as the vertebral column. Approximately 50% to 75% of patients treated with amphotericin B for nonmeningeal disease achieve a sustained remission, and therapy usually is curative in patients with infections localized strictly to skin and soft tissues without extensive abscess formation or tissue damage. The efficacy of local injection into joints or the peritoneum, as well as intraarticular or intradermal administration, remains poorly studied. Amphotericin B appears to be most efficacious when cell-mediated immunity is intact (as evidenced by a positive coccidioidin or spherulin skin test or low CF antibody titer). Controlled trials that document these clinical impressions are lacking, however.11,23
Meningeal Disease
Fluconazole has become the drug of choice for the treatment of coccidioidal meningitis. A minimum dose of 400 mg/day orally leads to a clinical response in most patients and obviates the need for intrathecal amphotericin B. Some clinicians will initiate therapy with 800 or 1,000 mg/day, and itraconazole dosages of 400 to 600 mg/day are comparably effective. It is also clear, however, that fluconazole only leads to remission rather than cure of the infections; thus suppressive therapy must be continued for life. Ketoconazole cannot be recommended routinely for the treatment of coccidioidal meningitis because of its poor CNS penetration following oral administration. Patients who do not respond to fluconazole or itraconazole therapy are candidates for intrathecal amphotericin B therapy with or without continuation of azole therapy. The intrathecal dose of amphotericin B ranges from 0.01 to 1.5 mg given at intervals ranging from daily to weekly. Therapy is initiated with a low dosage and is titrated upward as patient tolerance develops.11,23
CRYPTOCOCCOSIS
Epidemiology
Cryptococcosis is a noncontagious, systemic mycotic infection caused by the ubiquitous encapsulated soil yeast Cryptococcus, which is found in soil, particularly in pigeon droppings, although disease occurs throughout the world, even in areas where pigeons are absent. Infections caused by C. neoformans var. grubii (serotype A) are seen worldwide among immunocompromised hosts, followed by C. neoformans var. neoformans (serotype D). On the other hand, Cryptococcus gattii (serotypes B and C) is geographically more restricted and in contrast to C. neoformans, rarely infects immunosuppressed patients, is not associated with HIV infection, and the infections are more difficult to treat. C. gattii is not associated with birds; its main reservoir was thought to be limited to certain species of eucalyptus tree. Until recently, it was most common in tropical and subtropical areas, such as Australia, South America, Southeast Asia, and central Africa, with the highest incidence in Papua New Guinea and Northern Australia, although infections occur in nontropical areas such as North America and Europe. C. gattii emerged on Vancouver Island, British Columbia, Canada, in 1999, and subsequently spread to the Vancouver lower mainland, Washington state, and Oregon.24
Infection is acquired by inhalation of the organism. The incidence of cryptococcosis has risen dramatically in recent years, reflecting the increased numbers of immunocompromised patients, including those with malignancies, diabetes mellitus, chronic renal failure, and organ transplants and those receiving immunosuppressive agents. The AIDS epidemic also has contributed to the increased numbers of patients; cryptococcosis is the fourth most common infectious complication of AIDS and the second most common fungal pathogen. In most developed countries, widespread use of HAART has significantly decreased the incidence of cryptococcosis; however, the incidence and mortality of this infection are still extremely high in areas with limited access to HAART and a high incidence of HIV.25
Cell-mediated immunity appears to play a major role in host defense against infection with C. neoformans; 29% to 55% of patients with cryptococcal meningitis have a predisposing condition. Many patients with disseminated cryptococcosis demonstrate defects in cell-mediated immunity. The predilection of C. neoformans for the CNS appears to be caused by the lack of immunoglobulins and complement and the excellent growth medium afforded by CSF.25
Disease can remain localized in the lungs or can disseminate to other tissues, particularly the CNS, although the skin also can be affected. Hematogenous spread generally occurs in the immunocompromised host, although it also has been seen in individuals with intact immune systems.
Clinical Presentation of Cryptococcosis
Primary cryptococcosis in humans almost always occurs in the lungs, although the pulmonary focus usually produces a subclinical infection.23–28 Symptomatic infections usually are manifested by cough, rales, and shortness of breath that generally resolve spontaneously. Cryptococcus can present as part of an immune reconstitution inflammatory syndrome (IRIS), a paradoxical worsening of preexisting infectious processes following the initiation of HAART in HIV-infected individuals. In non-AIDS patients, the symptoms of cryptococcal meningitis are nonspecific. Headache, fever, nausea, vomiting, mental status changes, and neck stiffness generally are observed. Less common symptoms include visual disturbances (photophobia and blurred vision), papilledema, seizures, and aphasia. In AIDS patients, fever and headache are common, but meningismus and photophobia are much less common than in non-AIDS patients. Approximately 10% to 12% of AIDS patients have asymptomatic disease, similar to the rate observed in non-AIDS patients.25,27,28 Intracerebral mass lesions (cryptococcomas) are more common in C. gattii than in C. neoformans, presumably due to their different host immune responses.24
Laboratory Tests
With cryptococcal meningitis, the CSF opening pressure generally is elevated. There is a CSF pleocytosis (usually lymphocytes), leukocytosis, a decreased glucose concentration, and an elevated CSF protein concentration. There is also a positive cryptococcal antigen (detected by LA). The test is rapid, specific, and extremely sensitive, but false-negative results can occur. False-positive tests can result from cross-reactivity with rheumatoid factor and Trichosporon beigelii. C. neoformans can be detected in approximately 60% of patients by India ink smear of CSF, and it can be cultured in more than 96% of patients. Occasionally, large volumes of CSF are required to confirm the diagnosis.
The CSF parameters in patients with AIDS are similar to those seen in non-AIDS patients, with the exception of a decreased inflammatory response to the pathogen, resulting in a strikingly low number of leukocytes in CSF and extraordinarily high cryptococcal antigen titers.
TREATMENT
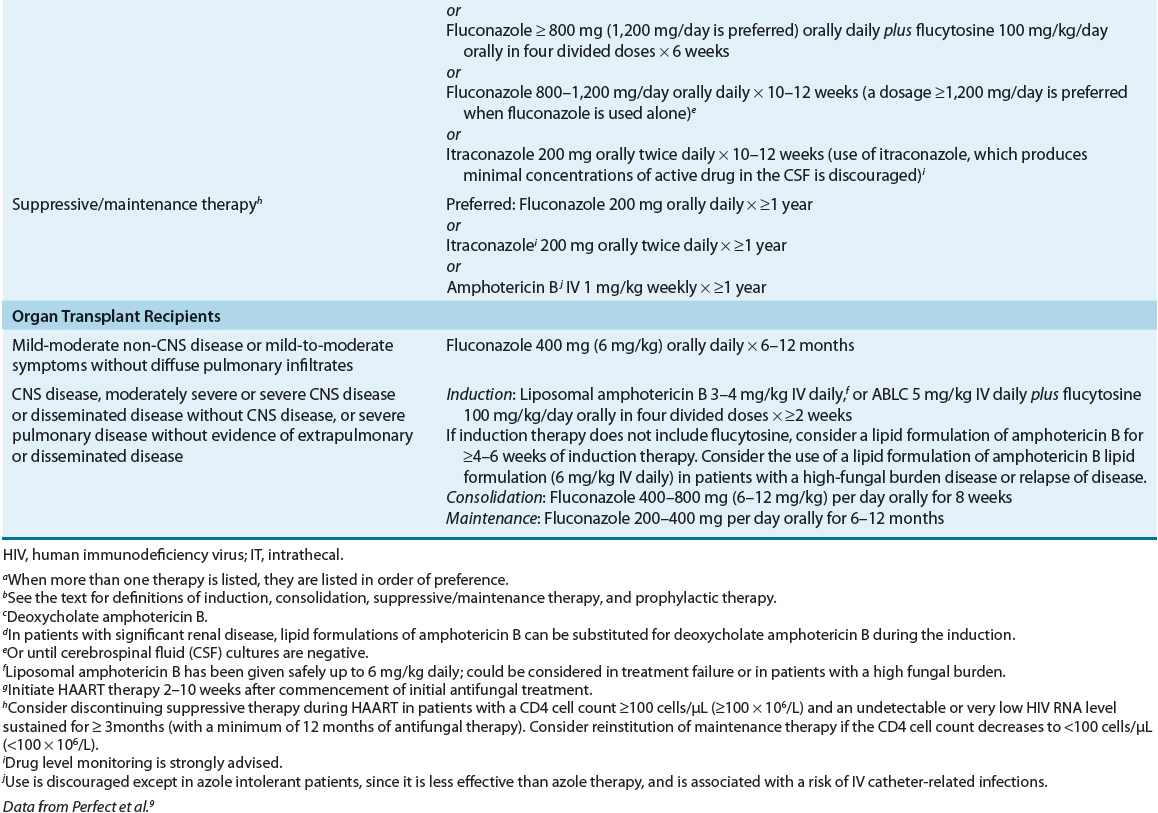
The choice of treatment for disease caused by C. neoformans depends on both the anatomic sites of involvement and the host’s immune status, and thus, treatment recommendations are divided into three specific risk groups: (a) HIV-infected individuals, (b) transplant recipients, and (c) non–HIV-infected and nontransplant hosts (Table 99–6).9 The management of cryptococcosis includes systemic antifungal therapy, control of elevated ICP, and supportive care. When possible, immune defects should be addressed. Despite the lack of randomized clinical trials, outcomes of treatment for CNS cryptococcosis (without mass lesions or hydrocephalus) appear to be similar for disease due to either C. neoformans or C. gattii, although no randomized clinical trials have been performed to address this.24
TABLE 99-6 Therapy of Cryptococcosisa,b