of single genes with a large effect on the phenotype. In alkaptonuria, Garrod observed that the amount of protein ingested was proportional to the darkness of urine and thus the amount of alkaptones excreted. Most individuals did not express this phenomenon, but asymptomatic carriers could be stressed with protein and excrete discernible amounts of alkaptones. Thus arose the “individualistic” concept that an individual’s genes controlled metabolism and that disease states were created by blocks in this metabolic flow that yielded accumulated precursors and deficient products.
and amino acids are determined from their butylester derivatives. In 2006, the American College of Medical Genetics (11) recommended a uniform panel of 29 disorders with primary targets and 25 secondary disorders. Forty-two disorders are screened for by MS/MS of dried blood spots, and all these disorders require rapid retrieval of the screen-positive newborn and urgent confirmation or denial of the screening results with immediate dietary intervention for the confirmed diagnosis. These metabolic conditions were selected through an iterative process among clinicians, laboratorians, and nutritionists. Selection was based on sufficient knowledge and availability of screening and confirmatory technology, the possibility for preventive intervention, and the probability that a screenpositive infant for one of these disorders would have a good outcome from the early intervention. Some of the disorders screened for, their MS/MS profiles, and the acute interventions required for a screen-positive infant are outlined in Table 69.1 (11, 12, 13, 14, 15, 16, 17, 18, 19, 20). The impaired enzymes are listed in Table 69.2 (15, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36). Because the initiation of emergency nutrition in the newborn changes with diagnostic confirmation and with increasing age, some recommended nutritional changes over the age of the child are listed in Table 69.3.
Enhancing anabolism and depressing catabolism. This combined approach involves the use of high-energy feedings and appropriate amino acid mixtures for aminoacidopathies and organic acidemias. An attempt should be made to prevent the physiologic catabolic state in the 4- to 14-day-old infant, and an anabolic state should be maintained throughout childhood. This therapeutic maneuver should be common to all inborn errors involving catabolic pathways. Caution must be exercised, however, to use high-energy feedings primarily during the acute presentation, but not on a long-term basis, to prevent overweight or obesity.
Correcting the primary imbalance in metabolic relationships. This correction involves using both dietary restriction to reduce accumulated substrates that are toxic and provision of products that may be deficient. For example, in phenylalanine hydroxylase (PAH) deficiency, PHE is restricted and TYR is supplemented.
Enhancing excretion of accumulated substances that are overproduced. The kidney can aid as a dialysis organ in removing accumulated toxic precursors. Maintaining diuresis with hydration is a critical component of therapy.
Providing alternative metabolic pathways to decrease accumulated toxic precursors in blocked reaction sequences. Many examples of this approach exist. For instance, the accumulated ammonia in enzyme defects of the urea cycle is reduced by removing nitrogen through administration of therapeutic amounts of phenylacetic acid (less noxious precursors, phenylbutyrate are used) to form phenylacetylglutamine from glutamine with the consequent loss of two nitrogen atoms in urine. Benzoic acid is also used to force glycine adducts of hippuric acid that result in the per mole urinary excretion of one nitrogen atom. Similarly, in isovaleric acidemia, innocuous isovalerylglycine (IVG) is formed from accumulating isovaleric acid (IVA) if supplemental glycine (GLY) is provided to drive the ubiquitous glycine-N-transacylase. IVG is then excreted in the urine. Betaine is used to drive methylation of homocysteine to methionine (MET) in cystathionine β-synthase (CβS) deficiency.
Using metabolic inhibitors to lower overproduced products. For example, allopurinol inhibits xanthine oxidase and decreases overproduction of uric acid in gout; lovastatin and compactin suppress hydroxymethylglutaryl -coenzyme A (CoA) reductase and reduce excess endogenous cholesterol biosynthesis in familial hypercholesterolemia; and 2-(2- nitro-4-trifluorome thylbenzoyl)-1,3-cyclohexanedione (NTBC) inhibits p-hydroxyphenylpyruvic acid dioxygenase (p-OHPPAD) and thus the toxic succinylacetone production in type I tyrosinemia.
Supplying products of blocked secondary pathways. In cystic fibrosis, the exocrine pancreas does not function in a normal manner to produce and secrete digestive enzymes. Administration of these pancreatic enzymes partially corrects this insufficiency and may prevent the sequelae of fat-soluble vitamin deficiency in the newborn and young child.
Stabilizing altered enzyme proteins. The rate of bio logic synthesis and degradation of holoenzymes depends on their structural conformation. In some holoenzymes, saturation by coenzyme increases their biologic halflife and thus overall enzyme activity at the equilibrium
produced by mutant proteins. This therapeutic mechanism is exemplified in PKU, homocystinuria, and MSUD. Tetrahydrobiopterin (BH4 [Kuvan]) is available as a drug for therapy of patients with non-PKU hyperphenylalaninemia to enhance PAH activity (37). Pharmacologic intake of vitamin B6 in homocystinuria or vitamin B1 in MSUD increases intracellular pyridoxal phosphate or thiamin pyrophosphate (TPP) and increases the specific activity of CβS or BCKAD complex, respectively (38, 39, 40, 41). Another approach is to provide chemical chaperones to stabilize mutant proteins. For example, excess intravenous infusion of D-galactose has increased defective α-galactosidase in the cardiac variant of Fabry disease (42).
Replacing deficient coenzymes. Various vitamindependent disorders are caused by blocks in coenzyme production and are “cured” by lifetime pharmacologic intake of a specific vitamin precursor. This mechanism presumably involves overcoming a partially impaired enzyme reaction by mass action. If reactions that are required to produce methylcobalamin (CH3-B12) or adenosylcobalamin are impaired, homocystinuria or methylmalonic aciduria (or both) will result. Daily intake of milligram quantities of vitamin B12 may cure both disorders (43). In biotinidase deficiency, the coenzyme biotin is not released from its covalently bound state. Reviews of “vitamin-dependency syndromes” have been published (39, 40, 41).
Artificially inducing enzyme production. If the structural gene or enzyme is intact, but suppressor, enhancer, or promoter elements are not functional, abnormal amounts of enzyme may be produced. It should be possible to “turn on” or “turn off” the structural gene and enable normal enzymatic production to occur. PAH activity has been induced in patients diagnosed with PKU when they received PHE loading for 3 days with intact protein (44, 45). Polyethylene glycol-coated phenylalanine ammonia lyase is undergoing clinical studies in patients with PKU to determine efficacy and allergenicity (46). In the acute porphyria of type I tyrosinemia, excessive σ-aminolevulinic acid (σ-ALA) production may be reduced by suppressing transcription of the σ-ALA synthase gene with excess glucose (GLU) and hematin (Figs. 69.1 and 69.2). In type I tyrosinemia, the overproduction of succinylacetone may be “turned off” by blocking an earlier enzyme p-OH-phenylpyruvate oxidase with the drug NBTC.
Replacing enzymes. Many attempts to replace deficient enzymes by plasma infusion and microencapsulation have been tried, with limited success. The use of polyethylene glycol coating of adenosine deaminase significantly prolonged the biologic half-life of this enzyme in treating severe combined immunodeficiency (47). The engineering of β- glucosidase with a high mannan receptor site enables intravenous use of
alglucerase (Ceredase) to treat type I Gaucher disease. Human α-galactosidase A replacement therapy in Fabry disease reverses substrate storage in the lysosome (42). Recombinant human α-glucosidase can prevent progression and improve cardiac and muscle function in Pompe disease (48). Polyethylene glycol-coated phenylalanine ammonia lyase is undergoing clinical studies in patients with PKU to determine efficacy and allergenicity (46).
Transplanting organs. Liver transplantation in a host of inherited metabolic disorders benefits systemic metabolism; the return of organ function replaces deficient enzyme activity (49, 50). Bone marrow and kidney transplantations are also of benefit.
Correcting the underlying defect in DNA so the body can manufacture its own functionally normal enzymes. The DNA for many proteins that are functionally deficient such as adenosine deaminase, hypoxanthine-guanine phosphoribosyl transferase, and ornithine transcarbamylase (OTC) and the lowdensity lipoprotein cholesterol receptor have been cloned, and retroviral constructs containing their cDNA have been transfected into dividing somatic
cells from affected individuals. Human gene therapy is currently contemplated for these inborn errors, although several technical problems in the toxicity of vectors, gene stability, and gene expression must be solved first. Other molecular approaches such as homologous recombination to correct mutant sequences or to inhibit RNA for dominant disorders producing antagonism to the normal allele are also future possibilities (51, 52).
Preventing absorption of a nutrient that is toxic in excess. Large neutral amino acids (LNAAs) free of PHE are available for preventing absorption of PHE from the intestinal tract and from passage across the blood-brain barrier (53).
TABLE 69.1 CORE AND SECONDARY TARGETS OF INBORN ERRORS OF METABOLISM WITH ONLINE MENDELIAN INHERITANCE IN MAN NUMBER RECOMMENDED FOR NEWBORN SCREENING, MARKER ANALYTES USED WITH TANDEM MASS SPECTROMETRY SCREENING, AND NUTRITION SUPPORT DURING DIAGNOSIS AND ACUTE THERAPY | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
TABLE 69.2 CHROMOSOMAL LOCATION, GENE SIZE, NUMBER OF MUTATIONS, TISSUE DISTRIBUTION OF GENES, AND GENOTYPE/PHENOTYPE CORRELATIONS | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
TABLE 69.3 APPROXIMATE DAILY REQUIREMENTS FOR SELECTED NUTRIENTSa BY INFANTS AND CHILDREN WITH SELECTED INHERITED DISORDERS OF AMINO ACID AND ORGANIC ACID METABOLISM | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
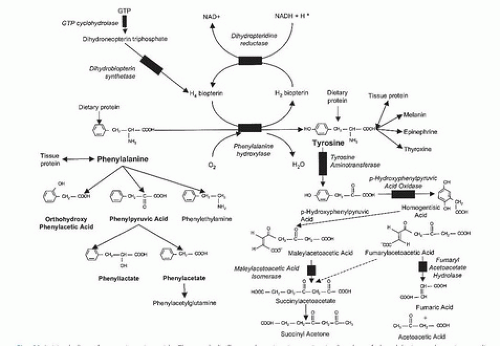 Fig. 69.1. Metabolism of aromatic amino acids. The metabolic flow and nutrient interaction in disorders of phenylalanine and tyrosine are diagrammed. The black bars represent impaired enzymes in biopterin biosynthesis, phenylketonuria, and tyrosinemia. GTP, guanosine triphosphate; NAD, nicotinamide adenine dinucleotide (NADH is the reduced form). |
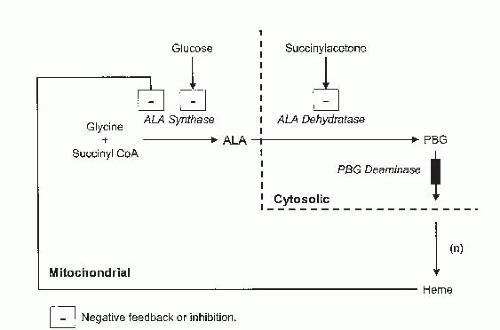 Fig. 69.2. Inhibition site in heme biosynthesis of relevance to diagnosis and treatment of tyrosinemia type I. The black bar represents the partial block in acute intermittent porphyria with resultant overproduction of σ-aminolevulinic acid (σ-ALA) and porphobilinogen (PBG) with decreased heme biosynthesis. In type I tyrosinemia, succinylacetone is produced and inhibits σ-ALA dehydratase with accumulation of σ-ALA alone, which is neurotoxic. σ-ALA accumulation can be reduced by addition of excess dietary glucose (GLU) and by hematin infusions that negatively control σ-ALA synthase at levels of both enzyme and gene expression. CoA, coenzyme A. |
hyperactivity. If not treated before 2 weeks of age, the metabolic imbalance produces mental retardation that is worsened with increasing time to treatment. The defect in metabolism in classic PKU is associated with less than 2% of the activity of normal PAH, and these classic mutations are now defined (62). The enzyme is expressed primarily in liver.
of mental retardation (77). With the present early infant discharge from the newborn nursery and the increase in breast-feeding, lower PHE concentrations of 121 to 242 µmol/L (2 to 4 mg/dL) are considered positive, and follow-up is initiated (78). Approximately 1 in 14,000 white newborns in the United States is affected with PKU (79), whereas 1 in 132,000 newborns in the black population is affected (80). For non-PKU hyperphenylalaninemia without TYR elevation, the estimated incidence is 1 in 48,000 for all newborns (79).
protein, is the primary protein source (16). Thus, recommendations for protein for nutrition support exceed the RDA. A mean protein intake 24% higher than the 1980 RDA for age was associated with greater PHE tolerance and growth in infants with PKU than was found when mean protein intake was near the RDA (89). Recommendations for energy and fluid intake (Table 69.4) (1, 90, 91, 92, 93) are the same as those for physiologically normal individuals (1).
TABLE 69.4 RECOMMENDED DIETARY INTAKES FOR MINERALS AND VITAMINS BY INFANTS AND CHILDREN | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
most are designed for patients older than 12 months of age, and some contain no minerals and vitamins and lead to deficiencies.
TABLE 69.5 SOURCE OF MEDICAL FOODS FOR INHERITED METABOLIC DISEASES | |
|---|---|
|
TABLE 69.6 SAMPLE DIET: PHENYLKETONURIA (2 WEEKS OF AGE), WEIGHT 3.25 kg | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
differentiating between ingestion of inadequate calories and excess PHE intake, respectively.
by the fetal blood-brain barrier (149). Intraneuronal PHE concentrations of 600 µmol interfere with brain development by one or more of the several previously described mechanisms, including abnormal oligodendroglial migration and myelin and other protein synthesis (150). Thus, it is extremely important to maintain normal plasma PHE concentrations in the reproductive-age woman with PKU before conception and throughout gestation. Surviving children of untreated women fail to grow and develop normally (151). In fact, Kirkman (152) predicted that if the fertility of these women is normal and they are not treated with dietary control of PHE intake, the incidence of PKU-related mental retardation could return to the prescreening level after only one generation.
TABLE 69.7 RECOMMENDED PHENYLALANINE, TYROSINE, PROTEIN, FAT, ESSENTIAL FATTY ACID, AND ENERGY INTAKES FOR PREGNANT WOMEN WITH PHENYLKETONURIA | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


