Chapter 10 Inflammatory diseases of the subcutaneous fat
Inflammatory diseases of the subcutaneous fat are a source of considerable confusion and often cause diagnostic difficulty to clinicians and pathologists alike. This stems in part from the use of classifications and clinical descriptions based upon time-honored but outdated literature.1–4 Inadequate biopsy specimens are also a source of considerable difficulty, particularly the punch biopsy specimen, which often yields no subcutaneous fat at all. Similarly, histological subdivision into diseases that affect the lobule and those that affect the septa is to some extent artifactual and sometimes unrewarding since most disorders affect both.2,3,5 There is also a somewhat monotonous clinical presentation, with most patients complaining of deep-seated, variably tender or painful nodules, often affecting the lower extremities.
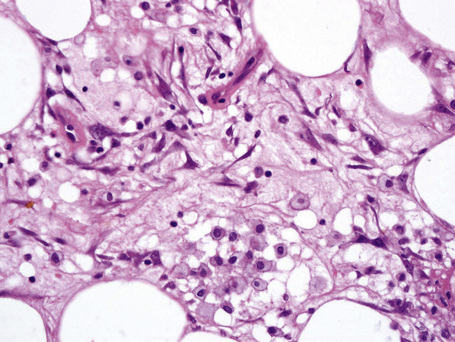
The subcutaneous fat has a limited repertoire of responses to noxious stimuli. Fat necrosis is a common manifestation of many forms of panniculitis and as a consequence there is often considerable histological overlap. Although there are many variants of fat necrosis – including enzymatic, crystalline, suppurative, hyalinizing and microcystic – lipophagic fat necrosis is the subtype most commonly encountered and is often a secondary feature in many forms of panniculitis (Fig. 10.1). This is characterized by a lobular infiltrate of histiocytes, xanthomatized cells, and foreign body giant cells, frequently accompanied by granulomata (Fig. 10.2).
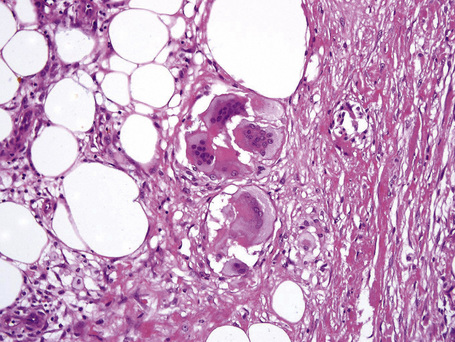
It is important to remember that the subcutaneous fat may be involved in a secondary manner, for example, in the vasculitides, the deep cutaneous fungal infections, by metastatic tumor, and following surgery or radiotherapy (Figs 10.3, 10.4). In this chapter the panniculitides are classified, where possible, on an etiological basis (Table 10.1).
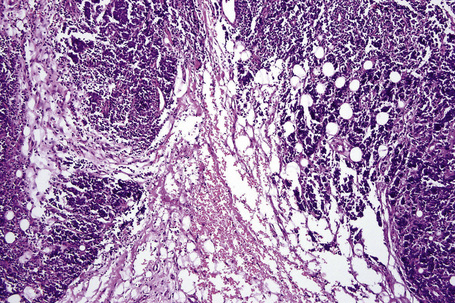
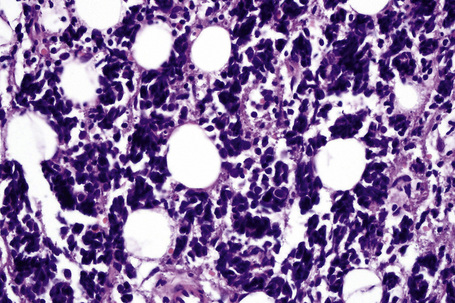
Table 10.1 Classification of panniculitis
Reproduced with permission from Peters, M.S. and Daniel Su, W.P. (1992). Panniculitis. Dermatologic Clinics, 10, 37–57.
There is considerable histological overlap in the various types of panniculitis and one must take into account all the clinical information before attempting to reach a definitive diagnosis. In patients in whom the diagnosis of panniculitis is suspected, a deep surgical incisional biopsy is essential (Fig. 10.5). The punch biopsy has no role whatsoever in the diagnosis of panniculitis.
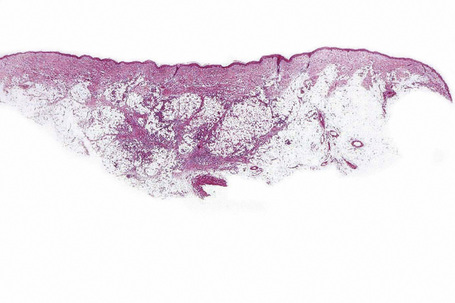
Fig. 10.5 Panniculitis: a deep surgical biopsy is essential in all cases where panniculitis is suspected.
Erythema nodosum
Clinical features
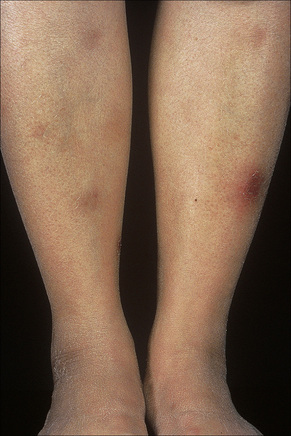
Erythema nodosum represents the commonest form of nodular panniculitis and is the prototype of septal panniculitis.1,2 It is, of course, a clinical syndrome rather than a specific disease in its own right, representing a complex of symptoms and signs with multiple and very variable etiologies.3,4 It typically affects young adults and shows a marked predilection for women (as high as 9:1 in some series). Children are only rarely affected.5 Patients present with a sudden onset of bright red, warm, tender nodules; these typically affect the anterior and lateral aspects of the lower legs, but the arms, face, calves, and trunk are occasionally involved (Figs 10.6, 10.7).6,7 Involvement of the soles of the feet is rare although it appears to be more often encountered in children.8,9 The lesions are usually multiple, bilateral, symmetrically distributed, elevated above the skin surface, and measure 1–15 cm in diameter.7 Ulceration and scarring are not features. Subsequently, the erythema fades to a bluish or livid hue and then to a yellow discoloration, reminiscent of a bruise (Fig. 10.8). The duration of the illness is 3–6 weeks. Patients sometimes also have pyrexia, malaise, and vague aches and pains in the joints. Laboratory findings may include a raised erythrocyte sedimentation rate (ESR), leukocytosis, and mild anemia.3
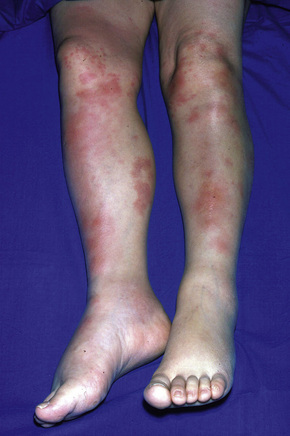
Fig. 10.6 Erythema nodosum: typical erythematous nodule on the shins of a young woman.
From the collection of the late N.P. Smith, MD, the Institute of Dermatology, London, UK.
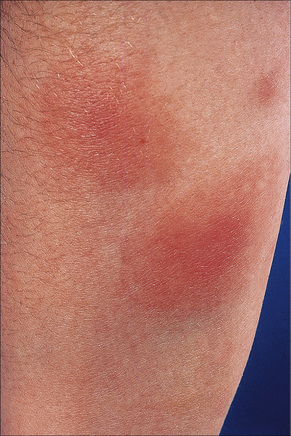
Fig. 10.7 Erythema nodosum: the lesions are raised and erythematous.
By courtesy of the Institute of Dermatology, London, UK.
Two clinical variants have been described.
Pathogenesis and histological features
The etiology and pathogenesis of erythema nodosum are unknown. Despite the very occasional finding of immunoreactants (IgM or IgG, and C3) in the blood vessel walls, an immune complex-mediated vasculitis is not considered likely.6 It is probable that erythema nodosum represents a non-specific hypersensitivity reaction that involves delayed hypersensitivity mechanisms in addition to a type 3 component.
There are many known associations. Although some are certainly of significance in the etiology of this dermatosis, many are probably coincidental (Table 10.2).15–28 In the earlier part of the twentieth century, tuberculosis was noted in up to 90% of adult patients with erythema nodosum, but this is now found in less than 1% of cases. Today, the more frequent associations include streptococcal infections, sarcoidosis, ulcerative colitis and Crohn’s disease, Sweet’s syndrome, Behçet’s disease, menstruation, pregnancy, estrogens and the oral contraceptive, cat scratch disease and various drug treatments (e.g., bromides, antibiotics, and sulfonamides). Other infectious conditions that have been described in association with erythema nodosum include cytomegalovirus, Epstein-Barr virus, parvovirus b19, Yersinia, Mycoplasma, Chlamydia, Brucella, Bartonella, Rickettsia, Helicobacter pylori, hepatitis B, atypical mycobacterial infections (e.g., swimming pool granuloma), Salmonella, Shigella, meningococcal septicemia, Q fever, leptospirosis, syphilis, human immunodeficiency virus (HIV), kerion, histoplasmosis, blastomycosis, amebiasis, ascaris, Chlamidophila pneumonia, and giardiasis.15,25–27,29–49 Additional drugs that have been implicated in the development of erythema nodosum include isotretinoin, interleukin (IL)-2, minocycline, thalidomide, echinacea, gold salts, hepatitis B vaccine, all-trans-retinoic acid, capecitabine, azathioprine, aromatase inhibitors, cabergoline, and lidocaine.16,50–62 Erythema nodosum has also been reported following a variety of malignancies including Hodgkin’s lymphoma, myelodysplastic syndrome, hairy cell leukemia, acute myeloid leukemia, acute myelomonocytic leukemia, diffuse large B-cell lymphoma, hypernephroma, non small cell lung carcinoma, pheochromocytoma, carcinoid tumor, hepatocellular carcinoma, carcinomas of the colon, pancreas and uterine cervix, and after radiotherapy.63–79 In 20–30% of patients no obvious cause is identified (idiopathic erythema nodosum).2 Erythema nodosum in renal transplant recipients can be related to infections, malignancies, drugs, inflammatory bowel disease or autoimmune diseases.80
Table 10.2 Erythema nodosum: etiology
| Streptococcus | Sarcoidosis |
| Tuberculosis | Sweet’s syndrome |
| Chlamydophila psittaci | Cat scratch disease |
| Crohn’s disease | Yersinia infection |
| Drugs | Ulcerative colitis |
| Behçet’s disease | Malignancy |
Erythema nodosum migrans seems to be particularly related to pregnancy, the oral contraceptive, streptococcal infection, and thyroid disease.10–12 Many cases, however, have no obvious associated predisposing factors or conditions.
Histologically, erythema nodosum represents the prototype of septal panniculitis. It is characterized by a combination of features, including vascular change, septal inflammation, hemorrhage, and a variable degree of acute or chronic panniculitis (Fig. 10.9). Although it is often said that erythema nodosum characteristically affects the septal component of the panniculus, it should be noted that there is not infrequently involvement of the lobule, in part or in whole, particularly if older lesions are biopsied. In the past, cases of the latter might have been diagnosed as Weber-Christian disease.
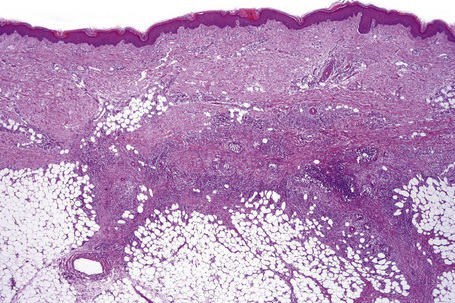
Frank vasculitis is only very exceptionally encountered. When present, it involves the small veins, and very occasionally medium-sized vessels within the connective tissue septa.81 It may be acute and necrotizing, associated with thrombosis and hemorrhage, or may manifest as chronic venular inflammation associated with endothelial cell swelling (Figs 10.10, 10.11). The overlying dermis typically shows a perivascular and periadnexal chronic inflammatory cell infiltrate.
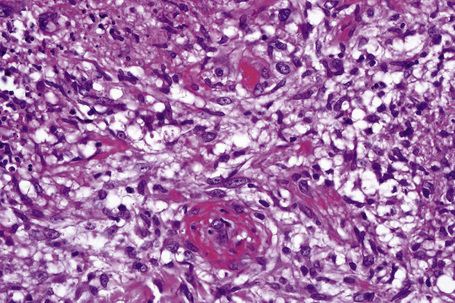
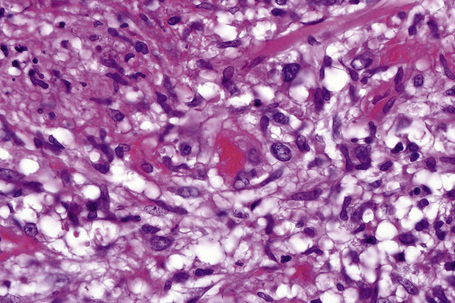
Fig. 10.11 Erythema nodosum: in this field there is a thrombosed venule associated with marked hemorrhage.
In the early stages, the septal inflammation may be acute, characterized by an infiltrate of neutrophil polymorphs, but this is soon replaced by lymphocytes and histiocytes (Figs 10.12–10.14).82,83 Eosinophils are sometimes found and rarely they can be conspicuous. Septal collections of histiocytes surrounding a cleftlike space (so-called Miescher’s radial granuloma) are said to be a characteristic feature, although they have been reported in Sweet’s syndrome, nodular vasculitis, and necrobiosis lipoidica (Figs 10.15–10.17).2,84,85 Further progression leads to the development of a frankly granulomatous infiltrate in which giant cells may be conspicuous. Coagulation and caseation-like necrosis are never seen in erythema nodosum (compare with nodular vasculitis below). Sometimes the connective tissue in the fibrous septa shows fibrinoid necrosis, and hemorrhage is almost invariably present (Figs 10.18–10.20).
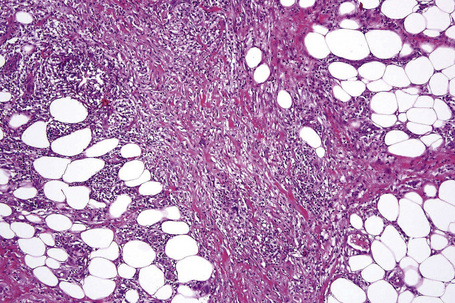
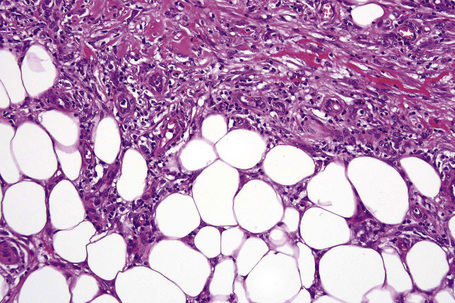
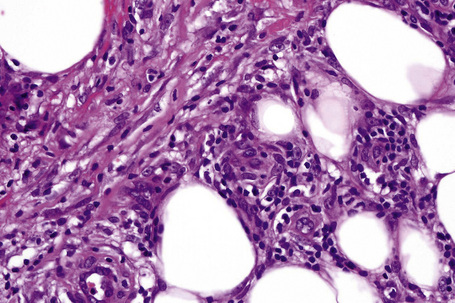
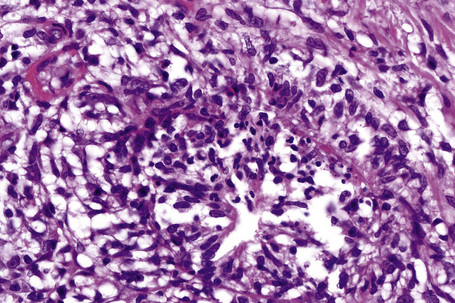
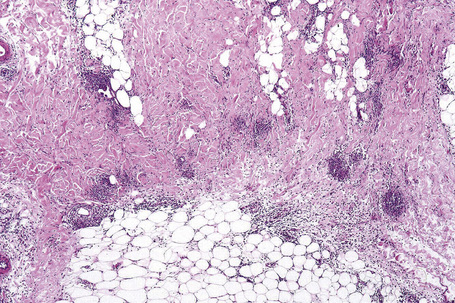
Fig. 10.16 Erythema nodosum: in this example multiple small granulomata are evident in the thickened septa.
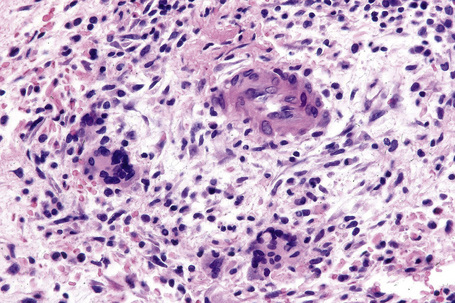
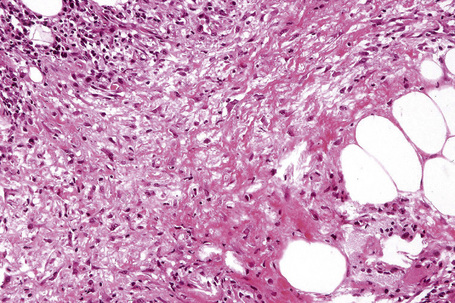
Fig. 10.18 Erythema nodosum: fibrinoid necrosis of the connective tissue septa is an occasional feature.
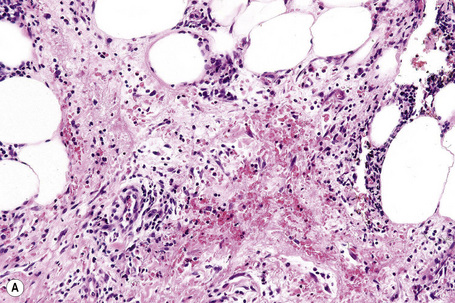
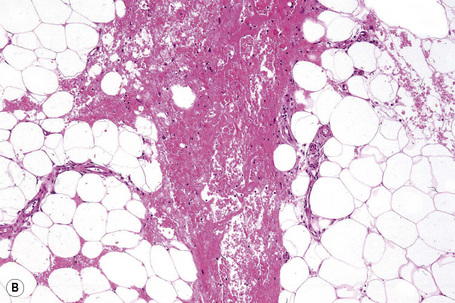
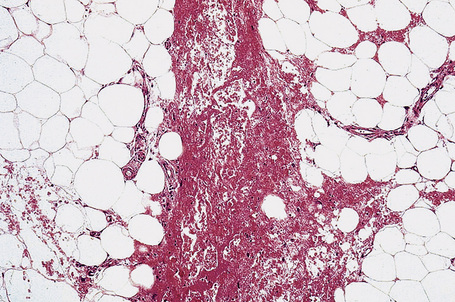
Characteristically, the septal infiltrate (lymphocytes, histiocytes, and granulomata) spills over to affect the periphery of the fat lobule to give a delicate lacy appearance, but fat necrosis is not usually present. On occasion, however, otherwise typical erythema nodosum may be associated with fat necrosis and a neutrophil inflammatory cell infiltrate (Fig. 10.21).81,86
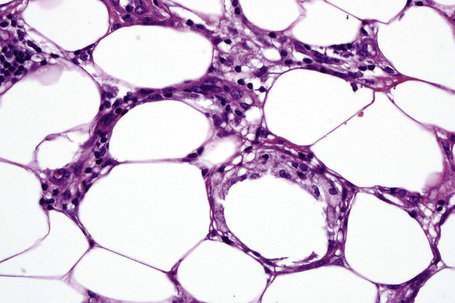
Erythema nodosum migrans is characterized by densely scarred and thickened interlobular septa accompanied by a conspicuous granulomatous infiltrate (Figs 10.22, 10.23). Numerous giant cells may be seen and often they form a palisade along the septal borders. Granulation tissue-like vascular proliferation is often a conspicuous feature. Vasculitis is absent and hemorrhage is not usually seen.
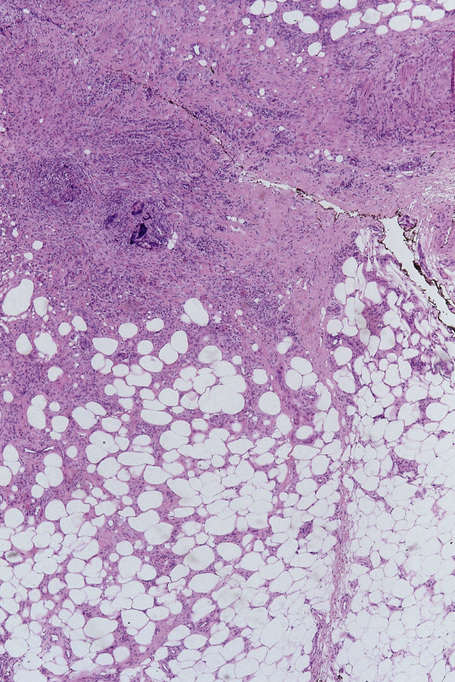
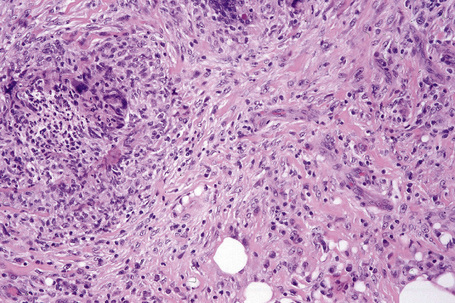
Fig. 10.23 Erythema nodosum migrans: close-up view showing granulomata and newly formed blood vessels.
In chronic erythema nodosum the histological changes are similar to, but usually milder, than those of the acute variant.11
Differential diagnosis
At scanning magnification, vasculitic processes affecting the septa of the subcutaneous fat may be mistaken for erythema nodosum. Occasionally, the features of leukocytoclastic vasculitis are seen within the septa in the absence of the more usual superficial dermal involvement.87 Such instances present as erythematous nodules, usually affecting the lower legs. Similarly, superficial thrombophlebitis presents within the subcutaneous fat septa. In this condition, however, the vein is the focus of the inflammatory process with associated thrombosis and there is little or no involvement of the lobule. Cutaneous polyarteritis nodosa affects muscular arteries within the lower dermis and subcutaneous fat septa and, therefore, should not be confused with erythema nodosum.87 Nephrogenic systemic fibrosis can rarely be associated with mild septal mononuclear cell infiltrates and granulomata, thereby simulating erythema nodosum.88
Erythema nodosum-like lesions in Behçet’s disease
Recurrent erythematous, tender, nodular lesions on the lower extremities (clinically reminiscent of erythema nodosum) are a common manifestation of Behçet’s disease.1–6 Erythema nodosum-like lesions develop in more than 40% of the patients during the course of the disease.7 Erythema nodosum-like lesions and superficial thrombophlebitis could represent predictive markers for visceral involvement.8
The fronts of the shins are most often affected but lesions may also occur on the arms, face, neck, and buttocks.2 Although histologically they have been described as showing erythema nodosum-like features, more commonly they are characterized by a lobular or mixed septal and lobular panniculitis associated with a neutrophil-rich infiltrate, neutrophilic vasculitis (affecting arterioles and venules), and associated fat necrosis.2,9 Less often, a lymphocytic vasculitis and, exceptionally, polyarteritis nodosa-like features are encountered. Miescher’s granulomata may sometimes be present.2,10
Weber-Christian disease
As originally defined by Christian in 1928 (relapsing febrile nodular nonsuppurative panniculitis), this disorder was characterized by recurrent attacks of fever associated with the development of subcutaneous tender nodules (particularly over the extremities), which were histologically characterized by the presence of nonsuppurative panniculitis which healed to leave a depressed scar.1–4 Lesions were said to affect mainly young white females, and although the lower extremities were predominantly affected, the upper extremities, buttocks, abdominal wall, breasts, and face could also be involved. Arthritis, arthralgias, and myalgias were often present.3 A systemic variant – which was potentially fatal and affected the intestines, mesentery, lungs, heart, and kidneys – was also recognized.3,5
Since 1928 there have been many case reports in the literature dealing with this so-called ‘specific disease’. In general, however, many of the (particularly earlier) studies used imprecise clinical and histological diagnostic criteria. Some were certainly examples of erythema nodosum. In the light of current knowledge of the panniculitides, many cases would now be reclassified. A Weber-Christian-like disease may be seen in erythema nodosum, factitial panniculitis, lupus panniculitis, pancreatic fat necrosis-associated panniculitis, α1-antitrypsin deficiency-associated panniculitis, connective tissue diseases, subcutaneous panniculitic T-cell lymphoma, and gamma-delta T-cell lymphoma.6–15 The term has also been applied to cases of infective panniculitis, and panniculitis following jejuno-ileal bypass surgery.16–19
It seems unlikely, therefore, that Weber-Christian disease represents a distinct entity in its own right. It is proposed, therefore, to take this opportunity to bury it once and for all. As suggested by Patterson, ‘a clinical diagnosis of Weber-Christian disease should signal the beginning of a search for the true cause of the disorder’.9 Likewise, the term Rothmann-Makai syndrome should be abandoned.20 More often than not it probably represents erythema nodosum.
α1-Antitrypsin deficiency-associated panniculitis
Clinical features
Deficiency of α1-antitrypsin is associated with a severe and particularly intractable form of panniculitis.1–14 Patients have recurrent episodes of painful or tender nodules which are particularly resistant to therapy. The disease shows a slight male predominance (3:2), and although a wide age range can be affected (7–73 years), most patients are in their fourth or fifth decade.5,7 Children, however, may occasionally be affected.5 The nodules, which are often precipitated by trauma, develop most often on the trunk and proximal extremities, but the buttocks, chest, back, and abdomen are sometimes also affected (Fig. 10.24). Occasionally, the disease spreads to the genitalia and involvement of the abdominal fat has been described.
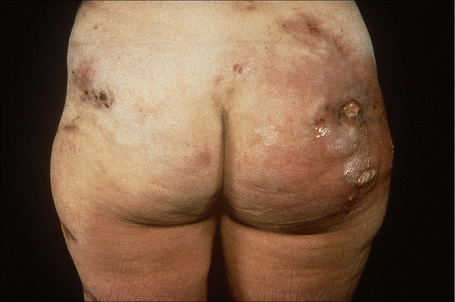
The nodules may be erythematous and are frequently associated with ulceration and the spontaneous discharge of clear or serosanguinous fluid.5 Deeply penetrating sinuses associated with liquefaction of the subcutaneous tissues are an important complication.
Fever is a common accompaniment and patients often have pulmonary problems including panacinar emphysema, chronic obstructive pulmonary disease, effusions, and embolic phenomena.5,15 Peripheral edema and anasarca are occasional manifestations. This is a particularly severe form of panniculitis, which has recently been successfully treated by the use of infusions of commercial α1-antitrypsin concentrate or liver transplantation.5,9,16,17 It is thought that many of the previously reported cases of Weber-Christian disease belong to this group.18
Panniculitis in association with α1-antitrypsin deficiency has been induced by cryosurgery,19 pregnancy, cesarean section delivery, and clarithromycin leak at the site of intravenous application.17,19–21 In one patient with the enzyme defect, Sweet’s syndrome was followed by the development of acquired cutis laxa (Marshall’s syndrome).22 An acquired α1-antitrypsin deficiency panniculitis following liver transplantation has been reported recently, which was successfully treated with re-transplantation of the liver.23
Pathogenesis and histological features
α1-Antitrypsin (a glycoprotein of hepatic derivation) is a serine protease inhibitor (PI) that greatly modifies the effects of proteolytic enzymes, accounting for at least 90% of serum proteolytic enzyme inhibition. In addition to antitrypsin inhibition, it is also responsible for inhibition of chymotrypsin, collagenase, elastase, factor VIII, and kallikrein.7 Its deficiency has been associated with panacinar emphysema, noninfective (neonatal and adult) hepatitis, and cirrhosis. More recently, associations have also been described with cutaneous vasculitis, atopic dermatitis, psoriasis, nodular prurigo, and cold urticaria.24 It has been proposed that absence of the protease inhibitor is associated with unrestrained complement activation with increased inflammatory cell activity, endothelial injury, and resultant autolytic tissue damage.25
Immunoglobulin (IgM) and complement (C3) have been identified in blood vessel walls in patients with this variant of panniculitis.6 The significance of this is uncertain.
The gene for α1-antitrypsin on chromosome 14 has in excess of 75 alleles and is inherited as an autosomal dominant.7 Deficiency occurs in between 1:3000 to 1:5000 of white North Americans.26 The MM genotype is most common and individuals with normal activity are coded PiMM. The ZZ genotype is associated with deficient α1-antitrypsin activity and the panniculitis is usually found in PiZZ individuals.27 Instances of panniculitis in PiMZ, PiSZ, PiMS, PiSS, and Null patients, however, have also been recorded.28–31 Panniculitis may also develop as a consequence of dysfunctional α1-antitrypsin.31,32 Recognition of this particular form is of importance as serum α1-antitrypsin levels are normal and therefore the diagnosis can easily be missed.
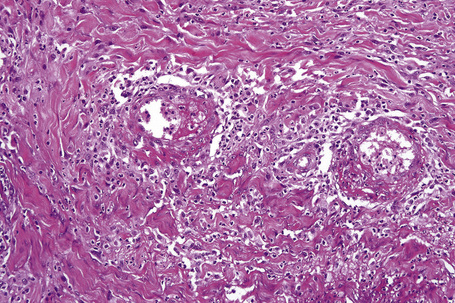
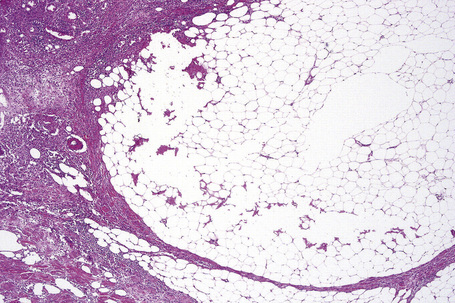
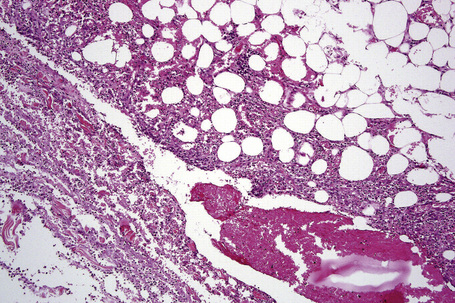
The earliest changes consist of necrosis of the connective tissue in the reticular dermis and septa of the subcutaneous fat accompanied by a neutrophil polymorph inflammatory cell infiltrate (Fig. 10.25).33 The histological features of an established lesion are those of a predominantly acute panniculitis (Fig. 10.26). The changes, which affect the septa and the paraseptal aspect of the lobule, are characteristically focal in nature. In acutely inflamed areas, large numbers of neutrophil polymorphs infiltrate the lobule. Fat necrosis is common and a characteristic feature is said to be the presence of normal fat adjacent to necrotic and inflamed fat (Figs 10.27, 10.28).6,34 Special stains often show fragmentation and loss of elastic tissue.6 Z-type α1-antitrypsin polymers have been demonstrated in the lesional as well as unaffected fatty tissue by immunohistochemistry in a single patient.35 Foci of hemorrhage associated with vascular thrombosis may be present, but there is no evidence of active vasculitis (Fig. 10.29).6 Elsewhere, a histiocytic infiltrate is conspicuous, involving both the deep vasculature and adjacent panniculus. Lipid-laden foamy macrophages are sometimes evident and multinucleate giant cells are occasionally found. Healing is by fibrous scarring.
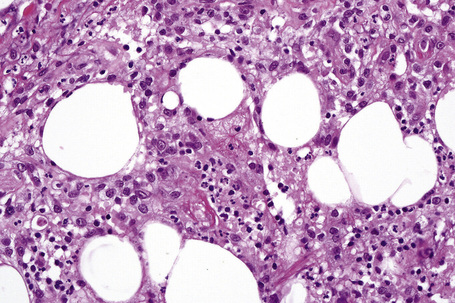
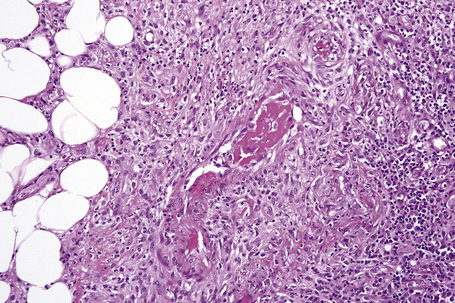
Factitial and traumatic panniculitis
Clinical features
By far the most common etiology is the subcutaneous injection of chemical substances including drugs, oily materials, and organic matter.1–4 Panniculitis has been described as a complication of morphine and tetanus antitoxoid injections. Similarly, repeated injections of pentazocine cause a characteristic woody fibrosis of the skin and subcutaneous fat accompanied by deeply penetrating ulcers and hyperpigmented halos.5–7 Pentazocine abuse has been described, particularly in members of the medical profession.5 There appears to be a relationship with a personal or family history of diabetes mellitus. It has been suggested that peripheral ischemia may be the pathogenetic link.7 A similar problem has recently been described following injections of the opioid ketobemidone.8
An important cause of factitial panniculitis is the repeated injection of oily materials including paraffin and liquid silicon (paraffinoma, sclerosing lipogranuloma, lipogranulomatous panniculitis) (Fig. 10.30).1,2,4,9–13 Sclerosing lipogranuloma was a condition usually seen in the male genitalia that developed as a consequence of the injection of paraffin oil and related compounds into the penis in the mistaken belief that this would enhance erections. Povidone (polyvinylpyrrolidone), a synthetic dispersing or suspending agent which has been used in both pharmaceutical products and hair sprays, may result in a particular characteristic histological variant of panniculitis.14,15 Associated features have included pulmonary lesions, lymphadenopathy, and hepatosplenomegaly. Organic substances that have been implicated in the etiology of factitial panniculitis include food matter, milk, and even feces.
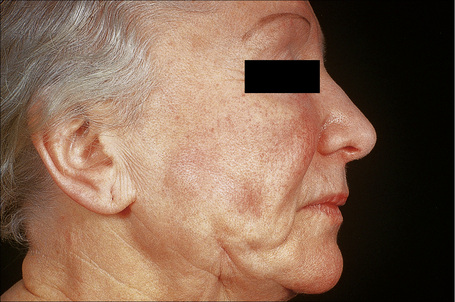
Fig. 10.30 Paraffinoma: note the infiltrated plaque with foci of retraction.
By courtesy of the Institute of Dermatology, London, UK.
Nodular cystic fat necrosis is a distinct posttraumatic lesion that is seen predominantly in adolescent boys and middle-aged women. Lesions, which are usually found on the legs, are often associated with a history of trauma.16
A number of therapeutic injections have been associated with the development of panniculitis including interferon-beta (IFN-β),17–21 glatiramer acetate,22–24 nadroparin-calcium25 and granulocyte colony stimulating factor.26 Aluminum granuloma may present as a panniculitis. Panniculitis has also been documented following vitamin K1 injections27. Two patients with factitial panniculitis caused by electroacupuncture have been recently described.28
Pathogenesis and histological features
The histological features of factitial panniculitis are not usually specific and depend to some extent upon the cause. In some instances, therefore, the changes are those of acute lobular inflammation associated with fat necrosis and a neutrophil polymorph infiltrate. In older lesions, mononuclear cells, lipid-laden histiocytes, and foreign body giant cells become predominant and sometimes the response becomes frankly granulomatous. On other occasions the septa may be primarily affected, thereby mimicking erythema nodosum. Calcification is occasionally evident.2 It is sometimes rewarding to view the sections with polarized light, as birefractile material may be identified, raising the possibility of the factitious nature of the condition.
Paraffinoma is characterized by the presence of round or oval spaces within the dermis and subcutaneous fat (‘Swiss cheese’ pattern) (Fig. 10.31); careful examination may reveal foamy histiocytes or giant cells lining the edges of these cystic cavities (Fig. 10.32).9 There is often associated dense fibrous scarring. Early lesions sometimes show a marked granulomatous component.9 Similar features have been described following a grease gun injury.29
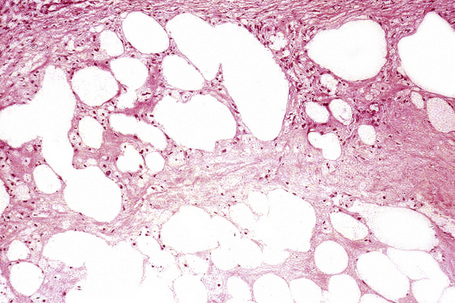
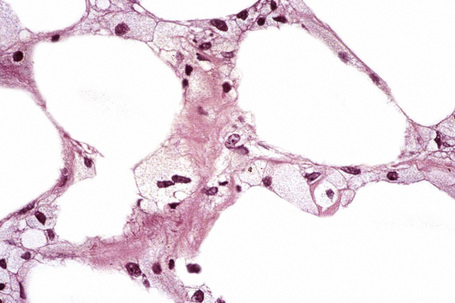
In panniculitis due to pentazocine abuse the histological features include dense dermal fibrosis accompanied by variable scarring of the subcutaneous fat.5 A ‘Swiss cheese’ appearance may be evident. Small-vessel thrombosis is frequently present.5
Povidone panniculitis is characterized by histiocytic accumulation of gray-blue, Congo red-positive foamy material accompanied by necrosis and hemorrhage.14
Lesions caused by blunt trauma show the features of an organizing hematoma. Granulomata and foci of hemosiderin pigment may additionally be present.30
Nodular cystic fat necrosis is thought to have an ischemic pathogenesis. Histologically, it is characterized by an encapsulated nodule of necrotic (anucleate) fat cells (Fig. 10.33).31,32 Variable inflammation is present.
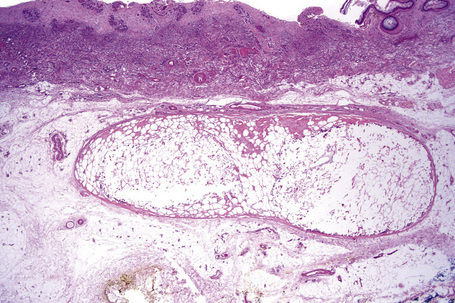
Fat necrosis with histiocytes (lipophages) and giant cells is a common histological finding in specimens taken from sites of previous surgery of the subcutaneous fat (or deeper). Zelickson and Winkelmann have described this as lipophagic panniculitis.31 Hemosiderin deposits are also commonly found and in many instances fragments of suture material may be identified.
The histological features of traumatic fat necrosis are not specific and are characterized by fat necrosis accompanied by a variable inflammatory cell infiltrate (Figs 10.34, 10.35). In early lesions this is predominantly composed of neutrophils, later replaced by lymphocytes and monocytes. Aggregates of lipophages are seen frequently and often the reaction becomes frankly granulomatous. Fat cysts are a common feature. With resolution, fibrosis takes place (Fig. 10.36). As evidence of the traumatic nature of the lesion, foci of hemosiderin deposition are not uncommon (Fig 10.37). Occasionally, the presence of fat necrosis is complicated by focal calcification (Fig. 10.38).
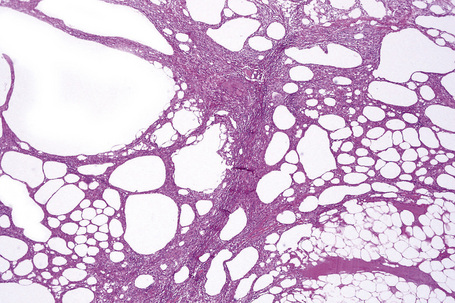
Fig. 10.34 Traumatic fat necrosis: there is intense lobular inflammation with septal fibrosis and hemorrhage.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


