389 | Relapsing Polychondritis |
Relapsing polychondritis is an uncommon disorder of unknown cause characterized by inflammation of cartilage predominantly affecting the ears, nose, and laryngotracheobronchial tree. Other manifestations include scleritis, neurosensory hearing loss, polyarthritis, cardiac abnormalities, skin lesions, and glomerulonephritis. Relapsing polychondritis has been estimated to have an incidence of 3.5 per million population per year. The peak age of onset is between the ages of 40 and 50 years, but relapsing polychondritis may affect children and the elderly. It is found in all races, and both sexes are equally affected. No familial tendency is apparent. A significantly higher frequency of HLA-DR4 has been found in patients with relapsing polychondritis than in healthy individuals. A predominant subtype allele(s) of HLA-DR4 was not found. Approximately 30% of patients with relapsing polychondritis will have another rheumatologic disorder, the most frequent being systemic vasculitis, followed by rheumatoid arthritis, and systemic lupus erythematosus (SLE). Nonrheumatic disorders associated with relapsing polychondritis include Hashimoto’s thyroiditis, primary biliary cirrhosis, and myelodysplastic syndrome (Table 389-1). In most cases, these disorders antedate the appearance of relapsing polychondritis, usually by months or years; however, in other instances, the onset of relapsing polychondritis can accompany disease presentation.
DISORDERS ASSOCIATED WITH RELAPSING POLYCHONDRITISa |
aSystemic vasculitis is the most common association, followed by rheumatoid arthritis and systemic lupus erythematosus.
Source: Modified from CJ Michet et al: Ann Intern Med 104:74, 1986.
PATHOLOGY AND PATHOPHYSIOLOGY
The earliest abnormality of hyaline and elastic cartilage noted histologically is a focal or diffuse loss of basophilic staining indicating depletion of proteoglycan from the cartilage matrix. Inflammatory infiltrates are found adjacent to involved cartilage and consist predominantly of mononuclear cells and occasional plasma cells. In acute disease, polymorphonuclear white cells may also be present. Destruction of cartilage begins at the outer edges and advances centrally. There is lacunar breakdown and loss of chondrocytes. Degenerating cartilage is replaced by granulation tissue and later by fibrosis and focal areas of calcification. Small loci of cartilage regeneration may be present. Immunofluorescence studies have shown immunoglobulins and complement at sites of involvement. Extracellular granular material observed in the degenerating cartilage matrix by electron microscopy has been interpreted to be enzymes, immunoglobulins, or proteoglycans.
Immunologic mechanisms play a role in the pathogenesis of relapsing polychondritis. The accumulating data strongly suggest that both humoral and cell-mediated immunity play an important role in the pathogenesis of relapsing polychondritis. Immunoglobulin and complement deposits are found at sites of inflammation. In addition, antibodies to type II collagen and to matrilin-1 and immune complexes are detected in the sera of some patients. The possibility that an immune response to type II collagen may be important in the pathogenesis is supported experimentally by the occurrence of auricular chondritis in rats immunized with type II collagen. Antibodies to type II collagen are found in the sera of these animals, and immune deposits are detected at sites of ear inflammation. Humoral immune responses to type IX and type XI collagen, matrilin-1, and cartilage oligomeric matrix protein have been demonstrated in some patients. In a study, rats immunized with matrilin-1 were found to develop severe inspiratory stridor and swelling of the nasal septum. The rats had severe inflammation with erosions of the involved cartilage, which was characterized by increased numbers of CD4+ and CD8+ T cells in the lesions. The cartilage of the joints and ear pinna was not involved. All had IgG antibodies to matrilin-1. Matrilin-1 is a noncollagenous protein present in the extracellular matrix in cartilage. It is present in high concentrations in the trachea and is also present in the nasal septum but not in articular cartilage. A subsequent study demonstrated serum anti-matrilin-1 antibodies in approximately 13% of patients with relapsing polychondritis; approximately 70% of these patients had respiratory symptoms. Cell-mediated immunity may also be operative in causing tissue injury, since lymphocyte transformation can be demonstrated when lymphocytes of patients are exposed to cartilage extracts. T cells specific for type II collagen have been found in some patients, and CD4+ T cells have been observed at sites of cartilage inflammation.
CLINICAL MANIFESTATIONS
The onset of relapsing polychondritis is frequently abrupt, with the appearance of one or two sites of cartilaginous inflammation. The pattern of cartilaginous involvement and the frequency of episodes vary widely among patients. Noncartilaginous presentations may also occur. Systemic inflammatory features such as fever, fatigue, and weight loss occur and may precede the clinical signs of relapsing polychondritis by several weeks. Relapsing polychondritis may go unrecognized for several months or even years in patients who only initially manifest intermittent joint pain and/or swelling, or who have unexplained eye inflammation, hearing loss, valvular heart disease, or pulmonary symptoms.
Auricular chondritis is the most frequent presenting manifestation of relapsing polychondritis, occurring in 40% of patients and eventually affecting about 85% of patients (Table 389-2). One or both ears are involved, either sequentially or simultaneously. Patients experience the sudden onset of pain, tenderness, and swelling of the cartilaginous portion of the ear (Fig. 389-1). This typically involves the pinna of the ears, sparing the earlobes because they do not contain cartilage. The overlying skin has a beefy red or violaceous color. Prolonged or recurrent episodes lead to cartilage destruction and result in a flabby or droopy ear. Swelling may close off the eustachian tube or the external auditory meatus, either of which can impair hearing. Inflammation of the internal auditory artery or its cochlear branch produces hearing loss, vertigo, ataxia, nausea, and vomiting. Vertigo is almost always accompanied by hearing loss.
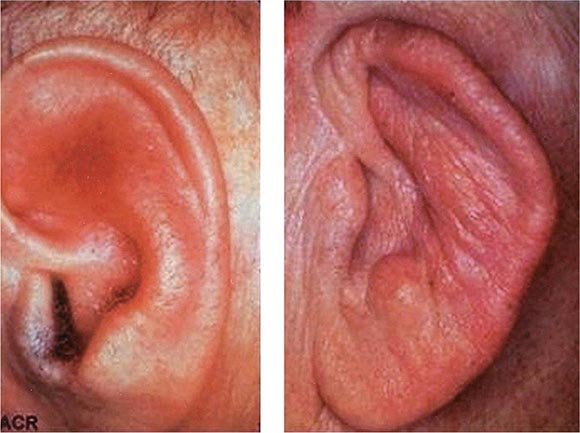
FIGURE 389-1 Left. The pinna is erythematous, swollen, and tender. Not shown is the ear lobule that is spared as there is no underlying cartilage. Right. The pinna is thickened and deformed. The destruction of the underlying cartilage results in a floppy ear. (Reprinted from the Clinical Slide Collection on the Rheumatic Diseases, ©1991, 1995, 1997, 1998, 1999. Used by permission of the American College of Rheumatology.)
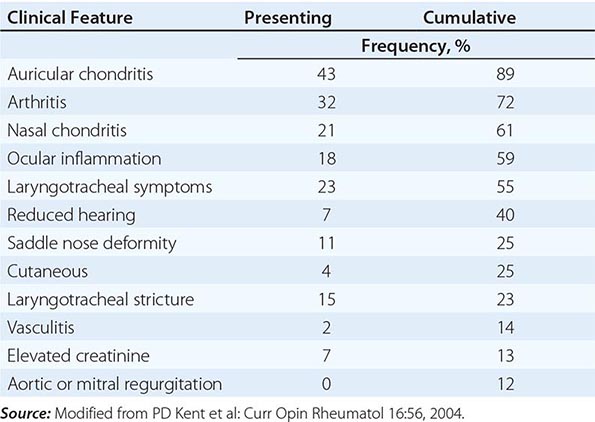
CLINICAL MANIFESTATIONS OF RELAPSING POLYCHONDRITIS |
Approximately 61% of patients will develop nasal involvement, with 21% having this at the time of presentation. Patients may experience nasal stuffiness, rhinorrhea, and epistaxis. The bridge of the nose and surrounding tissue become red, swollen, and tender and may collapse, producing a saddle nose deformity (Fig. 389-2). In some patients, nasal deformity develops insidiously without overt inflammation. Saddle nose is observed more frequently in younger patients, especially in women.
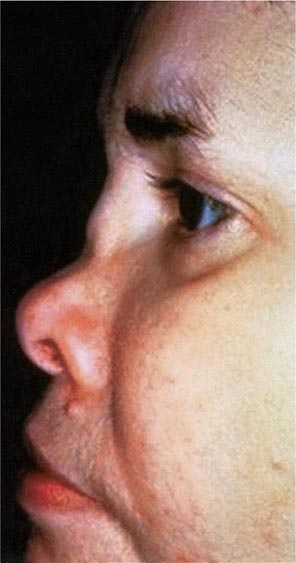
FIGURE 389-2 Saddle nose results from destruction and collapse of the nasal cartilage. (Reprinted from the Clinical Slide Collection on the Rheumatic Diseases, ©1991, 1995, 1997, 1998, 1999. Used by permission of the American College of Rheumatology.)
Joint involvement is the presenting manifestation in relapsing polychondritis in approximately one-third of patients and may be present for several months before other features appear. Eventually, more than one-half of the patients will have arthralgias or arthritis. The arthritis is usually asymmetric and oligo- or polyarticular, and it involves both large and small peripheral joints. An episode of arthritis lasts from a few days to several weeks and resolves spontaneously without joint erosion or deformity. Attacks of arthritis may not be temporally related to other manifestations of relapsing polychondritis. Joint fluid has been reported to be noninflammatory. In addition to peripheral joints, inflammation may involve the costochondral, sternomanubrial, and sternoclavicular cartilages. Destruction of these cartilages may result in a pectus excavatum deformity or even a flail anterior chest wall.
Eye manifestations occur in more than one-half of patients and include conjunctivitis, episcleritis, scleritis, iritis, uveitis, and keratitis. Ocular inflammation can be severe and visually threatening. Other manifestations include eyelid and periorbital edema, proptosis, optic neuritis, extraocular muscle palsies, retinal vasculitis, and renal vein occlusion.
Laryngotracheobronchial involvement occurs in ∼50% of patients and is among the most serious manifestations of relapsing polychondritis. Symptoms include hoarseness, a nonproductive cough, and tenderness over the larynx and proximal trachea. Mucosal edema, strictures, and/or collapse of laryngeal or tracheal cartilage may cause stridor and life-threatening airway obstruction necessitating tracheostomy. Involvement can extend into the lower airways resulting in tracheobronchomalacia. Collapse of cartilage in bronchi leads to pneumonia and, when extensive, to respiratory insufficiency.
Cardiac valvular regurgitation occurs in about 5–10% of patients and is due to progressive dilation of the valvular ring or to destruction of the valve cusps. Aortic regurgitation occurs in about 7% of patients, with the mitral and other heart valves being affected less often. Other cardiac manifestations include pericarditis, myocarditis, coronary vasculitis, and conduction abnormalities. Aneurysms of the proximal, thoracic, or abdominal aorta may occur even in the absence of active chondritis and occasionally rupture.
Renal disease occurs in about 10% of patients. The most common renal lesions include mesangial expansion or segmental necrotizing glomerulonephritis, which have been reported to have small amounts of electron-dense deposits in the mesangium where there is also faint deposition of C3 and/or IgG or IgM. Tubulointerstitial disease and IgA nephropathy have also been reported.
Approximately 25% of patients have skin lesions, which can include purpura, erythema nodosum, erythema multiforme, angioedema/urticaria, livedo reticularis, and panniculitis.
Features of vasculitis are seen in up to 25% of patients and can affect any size vessel. Large vessel vasculitis may present with aortic aneurysms, and medium vessel disease may affect the coronary, hepatic, mesenteric, or renal arteries or vessel supplying nerves. Skin vessel disease and involvement of the postcapillary venules can also occur. A variety of primary vasculitides have also been reported to occur in association with relapsing polychondritis (Chap. 385). One specific overlap is the “MAGIC” syndrome (mouth and genital ulcers with inflamed cartilage) in which patients present with features of both relapsing polychondritis and Behçet’s disease (Chap. 387).
LABORATORY FINDINGS AND DIAGNOSTIC IMAGING
There are no laboratory features that are diagnostic for relapsing polychondritis. Mild leukocytosis and normocytic, normochromic anemia are often present. Eosinophilia is observed in 10% of patients. The erythrocyte sedimentation rate and C-reactive protein are usually elevated. Rheumatoid factor and antinuclear antibody tests are occasionally positive in low titers, and complement levels are normal. Antibodies to type II collagen are present in fewer than one-half of the patients and are not specific. Circulating immune complexes may be detected, especially in patients with early active disease. Elevated levels of γ globulin may be present. Antineutrophil cytoplasmic antibodies (ANCA), either cytoplasmic (cANCA) or perinuclear (pANCA), are found in some patients with active disease. However, on target antigen–specific testing, there are only occasional reports of positive myeloperoxidase-ANCA, and proteinase 3-ANCA are very rarely found in relapsing polychondritis.
The upper and lower airways can be evaluated by imaging techniques such as computed tomography and magnetic resonance imaging (MRI). Bronchoscopy provides direct visualization of the airways but can be a high-risk procedure in patients with airway compromise. Pulmonary function testing with flow-volume loops can show inspiratory and/or expiratory obstruction. Imaging can also be useful to detect extracartilaginous disease. The chest film may show widening of the ascending or descending aorta due to an aneurysm, and cardiomegaly when aortic insufficiency is present. MRI can assess aortic aneurysmal dilatation. Electrocardiography and echocardiography can be useful in further evaluating for cardiac features of disease.
DIAGNOSIS
Diagnosis is based on recognition of the typical clinical features. Biopsies of the involved cartilage from the ear, nose, or respiratory tract will confirm the diagnosis but are only necessary when clinical features are not typical. Diagnostic criteria were suggested in 1976 by McAdam et al and modified by Damiani and Levine in 1979. These criteria continue to be generally used in clinical practice. McAdam et al proposed the following: (1) recurrent chondritis of both auricles; (2) nonerosive inflammatory arthritis; (3) chondritis of nasal cartilage; (4) inflammation of ocular structures, including conjunctivitis, keratitis, scleritis/episcleritis, and/or uveitis; (5) chondritis of the laryngeal and/or tracheal cartilages; and (6) cochlear and/or vestibular damage manifested by neurosensory hearing loss, tinnitus, and/or vertigo. The diagnosis is certain when three or more of these features are present along with a positive biopsy from the ear, nasal, or respiratory cartilage. Damiani and Levine later suggested that the diagnosis could be made when one or more of the above features and a positive biopsy were present, when two or more separate sites of cartilage inflammation were present that responded to glucocorticoids or dapsone, or when three or more of the above features were present.
The differential diagnosis of relapsing polychondritis is centered around its sites of clinical involvement. Patients with granulomatosis with polyangiitis (Wegener’s) may have a saddle nose and tracheal involvement but can be distinguished by the primary inflammation occurring in the mucosa at these sites, the absence of auricular involvement, and the presence of pulmonary parenchymal disease. Patients with Cogan’s syndrome have interstitial keratitis and vestibular and auditory abnormalities, but this syndrome does not involve the respiratory tract or ears. Reactive arthritis may initially resemble relapsing polychondritis because of oligoarticular arthritis and eye involvement, but it is distinguished in time by the appearance of urethritis and typical mucocutaneous lesions and the absence of nose or ear cartilage involvement. Rheumatoid arthritis may initially suggest relapsing polychondritis because of arthritis and eye inflammation. The arthritis in rheumatoid arthritis, however, is erosive and symmetric. In addition, rheumatoid factor titers are usually high compared with those in relapsing polychondritis, and anti-cyclic citrullinated peptide is usually not seen. Bacterial infection of the pinna may be mistaken for relapsing polychondritis but differs by usually involving only one ear, including the earlobe. Auricular cartilage may also be damaged by trauma or frostbite. Nasal destructive disease and auricular abnormalities can also be seen in patients using cocaine adulterated with levamisole. Ear involvement in this setting differs from relapsing polychondritis by typically manifesting as purpuric plaques with necrosis extending to the pinna, which does not contain cartilage.
PATIENT OUTCOME, PROGNOSIS, AND SURVIVAL
The course of relapsing polychondritis is highly variable. Some patients experience inflammatory episodes lasting from a few days to several weeks that then subside spontaneously or with treatment. Attacks may recur at intervals varying from weeks to months. In other patients, the disease has a chronic, smoldering course that may be severe. In one study, the 5-year estimated survival rate was 74% and the 10-year survival rate was 55%. In contrast to earlier series, only about one-half of the deaths could be attributed to relapsing polychondritis or complications of treatment. Airway complications accounted for only 10% of all fatalities. In general, patients with more widespread disease have a worse prognosis.
ACKNOWLEDGMENT
This chapter represents a revised version of the text authored by Dr. Bruce C. Gilliland that appeared in previous editions of Harrison’s Principles of Internal Medicine. Dr. Gilliland passed away on February 17, 2007, and had been a contributor to Harrison’s since the 11th edition.
390 | Sarcoidosis |
DEFINITION
Sarcoidosis is an inflammatory disease characterized by the presence of noncaseating granulomas. The disease is often multisystem and requires the presence of involvement in two or more organs for a specific diagnosis. The finding of granulomas is not specific for sarcoidosis, and other conditions known to cause granulomas must be ruled out. These conditions include mycobacterial and fungal infections, malignancy, and environmental agents such as beryllium. Although sarcoidosis can affect virtually every organ of the body, the lung is most commonly affected. Other organs commonly affected are the liver, skin, and eye. The clinical outcome of sarcoidosis varies, with remission occurring in over one-half of patients within a few years of diagnosis; however, the remaining patients may develop a chronic disease that lasts for decades.
ETIOLOGY
Despite multiple investigations, the cause of sarcoidosis remains unknown. Currently, the most likely etiology is an infectious or noninfectious environmental agent that triggers an inflammatory response in a genetically susceptible host. Among the possible infectious agents, careful studies have shown a much higher incidence of Propionibacter acnes in the lymph nodes of sarcoidosis patients compared to controls. An animal model has shown that P. acnes can induce a granulomatous response in mice similar to sarcoidosis. Others have demonstrated the presence of a mycobacterial protein (Mycobacterium tuberculosis catalase-peroxidase [mKatG]) in the granulomas of some sarcoidosis patients. This protein is very resistant to degradation and may represent the persistent antigen in sarcoidosis. Immune response to this and other mycobacterial proteins has been documented by another laboratory. These studies suggest that a mycobacterium similar to M. tuberculosis could be responsible for sarcoidosis. The mechanism exposure/infection with such agents has been the focus of other studies. Environmental exposures to insecticides and mold have been associated with an increased risk for disease. In addition, health care workers appear to have an increased risk. Also, sarcoidosis in a donor organ has occurred after transplantation into a sarcoidosis patient. Some authors have suggested that sarcoidosis is not due to a single agent but represents a particular host response to multiple agents. Some studies have been able to correlate the environmental exposures to genetic markers. These studies have supported the hypothesis that a genetically susceptible host is a key factor in the disease.
INCIDENCE, PREVALENCE, AND GLOBAL IMPACT
![]() Sarcoidosis is seen worldwide, with the highest prevalence reported in the Nordic population. In the United States, the disease has been reported more commonly in African Americans than whites, with the ratio of African Americans to whites ranging from 3:1 to 17:0. Women appear to be slightly more susceptible than men. The higher incidence in African Americans may have been influenced by the fact that African Americans seem to develop more extensive and chronic pulmonary disease. Because most sarcoidosis clinics are run by pulmonologists, a selection bias may have occurred. Worldwide, the prevalence of the disease varies from 20–60 per 100,000 for many groups such as Japanese, Italians, and American whites. Higher rates occur in Ireland and Nordic countries. In one closely observed community in Sweden, the lifetime risk for developing sarcoidosis was 3%.
Sarcoidosis is seen worldwide, with the highest prevalence reported in the Nordic population. In the United States, the disease has been reported more commonly in African Americans than whites, with the ratio of African Americans to whites ranging from 3:1 to 17:0. Women appear to be slightly more susceptible than men. The higher incidence in African Americans may have been influenced by the fact that African Americans seem to develop more extensive and chronic pulmonary disease. Because most sarcoidosis clinics are run by pulmonologists, a selection bias may have occurred. Worldwide, the prevalence of the disease varies from 20–60 per 100,000 for many groups such as Japanese, Italians, and American whites. Higher rates occur in Ireland and Nordic countries. In one closely observed community in Sweden, the lifetime risk for developing sarcoidosis was 3%.
Sarcoidosis often occurs in young, otherwise healthy adults. It is uncommon to diagnose the disease in someone under age 18. However, it has become clear that a second peak in incidence develops around age 60. In a study of >700 newly diagnosed sarcoidosis patients in the United States, one-half of the patients were ≥40 years at the time of diagnosis.
Although most cases of sarcoidosis are sporadic, a familial form of the disease exists. At least 5% of patients with sarcoidosis will have a family member with sarcoidosis. Sarcoidosis patients who are Irish or African American seem to have a two to three times higher rate of familial disease.
PATHOPHYSIOLOGY AND IMMUNOPATHOGENESIS
The granuloma is the pathologic hallmark of sarcoidosis. A distinct feature of sarcoidosis is the local accumulation of inflammatory cells. Extensive studies in the lung using bronchoalveolar lavage (BAL) have demonstrated that the initial inflammatory response is an influx of T helper cells. In addition, there is an accumulation of activated monocytes. Figure 390-1 is a proposed model for sarcoidosis. Using the HLA-CD4 complex, antigen-presenting cells present an unknown antigen to the helper T cell. Studies have clarified that specific HLA haplotypes such as HLA-DRB1*1101 are associated with an increased risk for developing sarcoidosis. In addition, different HLA haplotypes are associated with different clinical outcomes.
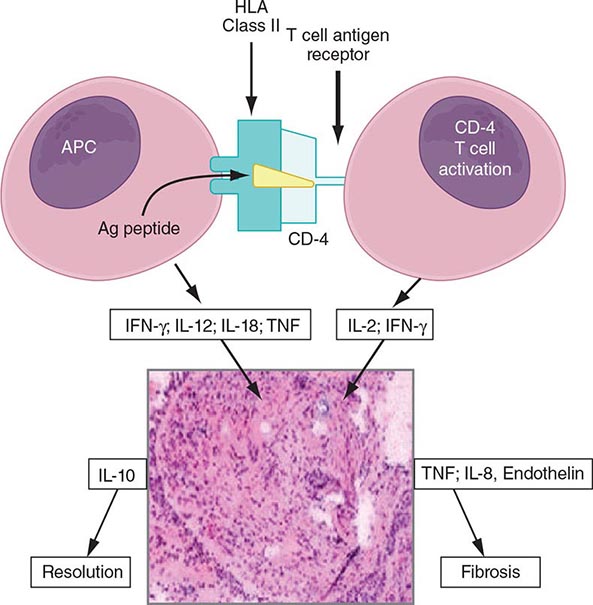
FIGURE 390-1 Schematic representation of initial events of sarcoidosis. The antigen-presenting cell and helper T cell complex leads to the release of multiple cytokines. This forms a granuloma. Over time, the granuloma may resolve or lead to chronic disease, including fibrosis. APC, antigen-presenting cell; HLA, human leukocyte antigen; IFN, interferon; IL, interleukin; TNF, tumor necrosis factor.
The macrophage/helper T cell cluster leads to activation with the increased release of several cytokines. These include interleukin (IL)-2 released from the T cell and interferon γ and tumor necrosis factor (TNF) released by the macrophage. The T cell is a necessary part of the initial inflammatory response. In advanced, untreated HIV infection, patients who lack helper T cells rarely develop sarcoidosis. In contrast, several reports confirm that sarcoidosis becomes unmasked as HIV-infected individuals receive antiretroviral therapy, with subsequent restoration of their immune system. In contrast, treatment of established pulmonary sarcoidosis with cyclosporine, a drug that downregulates helper T cell responses, seems to have little impact on sarcoidosis.
The granulomatous response of sarcoidosis can resolve with or without therapy. However, in at least 20% of patients with sarcoidosis, a chronic form of the disease develops. This persistent form of the disease is associated with the secretion of high levels of IL-8. Also, studies have reported that patients with this chronic form of disease release excessive amounts of TNF in areas of inflammation. Specific gene signatures have been associated with more severe disease, such as cardiac, neurologic, and fibrotic pulmonary disease.
At diagnosis the natural history of the disease may be difficult to predict. One form of the disease, Löfgren’s syndrome, consists of erythema nodosum and hilar adenopathy on chest roentgenogram. In some cases, periarticular arthritis may be identified without erythema nodosum. Löfgren’s syndrome is associated with a good prognosis, with >90% of patients experiencing disease resolution within 2 years. Recent studies have demonstrated that the HLA-DRB1*03 was found in two-thirds of Scandinavian patients with Löfgren’s syndrome. More than 95% of those patients who were HLA-DRB1*03 positive had resolution of their disease within 2 years, whereas nearly one-half of the remaining patients had disease for more than 2 years. It remains to be determined whether these observations can be applied to a non-Scandinavian population.
CLINICAL MANIFESTATIONS
The presentation of sarcoidosis ranges from patients who are asymptomatic to those with organ failure. It is unclear how often sarcoidosis is asymptomatic. In countries where routine chest roentgenogram screening is performed, 20–30% of pulmonary cases are detected in asymptomatic individuals. The inability to screen for other asymptomatic forms of the disease would suggest that as many as one-third of sarcoidosis patients are asymptomatic.
Respiratory complaints including cough and dyspnea are the most common presenting symptoms. In many cases, the patient presents with a 2- to 4-week history of these symptoms. Unfortunately, due to the nonspecific nature of pulmonary symptoms, the patient may see physicians for up to a year before a diagnosis is confirmed. For these patients, the diagnosis of sarcoidosis is usually only suggested when a chest roentgenogram is performed.
Symptoms related to cutaneous and ocular disease are the next two most common complaints. Skin lesions are often nonspecific. However, because these lesions are readily observed, the patient and treating physician are often led to a diagnosis. In contrast to patients with pulmonary disease, patients with cutaneous lesions are more likely to be diagnosed within 6 months of symptoms.
Nonspecific constitutional symptoms include fatigue, fever, night sweats, and weight loss. Fatigue is perhaps the most common constitutional symptom that affects these patients. Given its insidious nature, patients are usually not aware of the association with their sarcoidosis until their disease resolves.
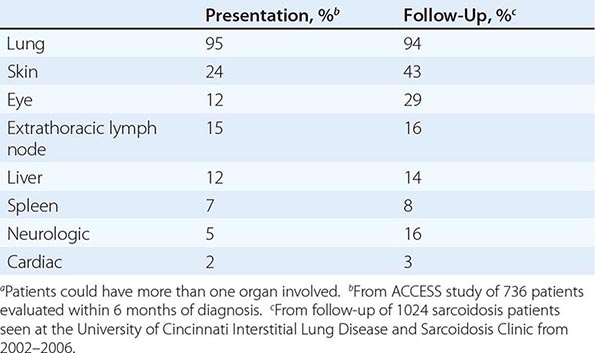
The overall incidence of sarcoidosis at the time of diagnosis and eventual common organ involvement are summarized in Table 390-1. Over time, skin, eye, and neurologic involvement seem more apparent. In the United States, the frequency of specific organ involvement appears to be affected by age, race, and gender. For example, eye disease is more common among African Americans. Under the age of 40, it occurs more frequently in women. However, in those diagnosed over the age of 40, eye disease is more common in men.
FREQUENCY OF COMMON ORGAN INVOLVEMENT AND LIFETIME RISKa |
LUNG
Lung involvement occurs in >90% of sarcoidosis patients. The most commonly used method for detecting lung disease is still the chest roentgenogram. Figure 390-2 illustrates the chest roentgenogram from a sarcoidosis patient with bilateral hilar adenopathy. Although the computed tomography (CT) scan has changed the diagnostic approach to interstitial lung disease, the CT scan is not usually considered a monitoring tool for patients with sarcoidosis. Figure 390-3 demonstrates some of the characteristic CT features, including peribronchial thickening and reticular nodular changes, which are predominantly subpleural. The peribronchial thickening seen on CT scan seems to explain the high yield of granulomas from bronchial biopsies performed for diagnosis.
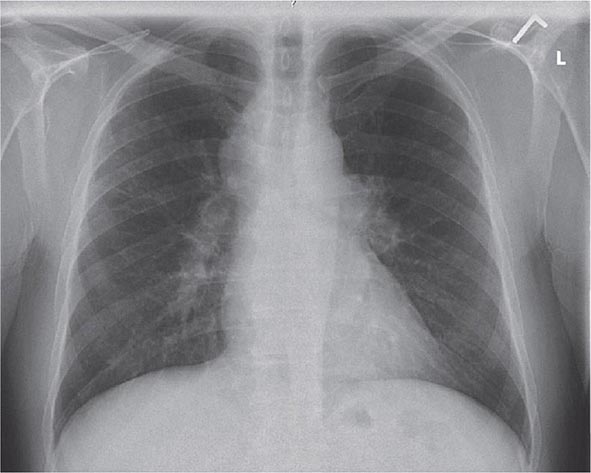
FIGURE 390-2 Posterior-anterior chest roentgenogram demonstrating bilateral hilar adenopathy, stage 1 disease.
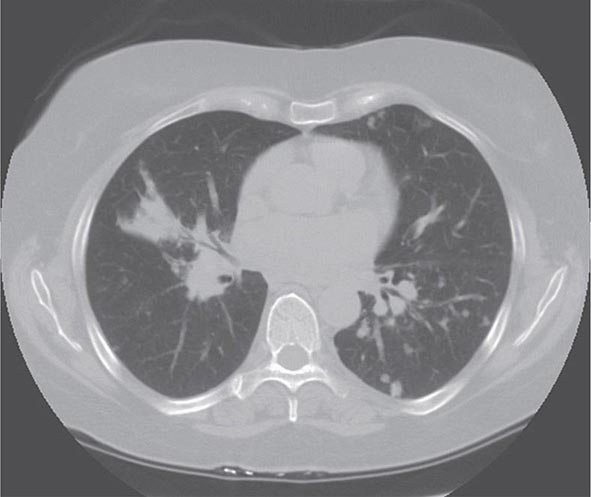
FIGURE 390-3 High-resolution computed tomography scan of chest demonstrating patchy reticular nodularity, including areas of confluence.
Although the CT scan is more sensitive, the standard scoring system described by Scadding in 1961 for chest roentgenograms remains the preferred method of characterizing the chest involvement. Stage 1 is hilar adenopathy alone (Fig. 390-2), often with right paratracheal involvement. Stage 2 is a combination of adenopathy plus infiltrates, whereas stage 3 reveals infiltrates alone. Stage 4 consists of fibrosis. Usually the infiltrates in sarcoidosis are predominantly an upper lobe process. Only in a few noninfectious diseases is an upper lobe predominance noted. In addition to sarcoidosis, the differential diagnosis of upper lobe disease includes hypersensitivity pneumonitis, silicosis, and Langerhans cell histiocytosis. For infectious diseases, tuberculosis and Pneumocystis pneumonia can often present as upper lobe diseases.
Lung volumes, mechanics, and diffusion are all useful in evaluating interstitial lung diseases such as sarcoidosis. The diffusion of carbon monoxide (DLCO) is the most sensitive test for an interstitial lung disease. Reduced lung volumes are a reflection of the restrictive lung disease seen in sarcoidosis. However, a third of the patients presenting with sarcoidosis still have lung volumes within the normal range, despite abnormal chest roentgenograms and dyspnea.
Approximately one-half of sarcoidosis patients present with obstructive disease, reflected by a reduced ratio of forced vital capacity expired in 1 second (FEV1/FVC). Cough is a very common symptom. Airway involvement causing varying degrees of obstruction underlies the cough in most sarcoidosis patients. Airway hyperreactivity, as determined by methacholine challenge, will be positive in some of these patients. A few patients with cough will respond to traditional bronchodilators as the only form of treatment. In some cases, high-dose inhaled glucocorticoids alone are useful. Airway obstruction can be due to large airway stenosis, which can become fibrotic and unresponsive to anti-inflammatory therapy.
Pulmonary arterial hypertension is reported in at least 5% of sarcoidosis patients. Either direct vascular involvement or the consequence of fibrotic changes in the lung can lead to pulmonary arterial hypertension. In sarcoidosis patients with end-stage fibrosis awaiting lung transplant, 70% will have pulmonary arterial hypertension. This is a much higher incidence than that reported for other fibrotic lung diseases. In less advanced, but still symptomatic, patients, pulmonary arterial hypertension has been noted in up to 50% of the cases. Because sarcoidosis-associated pulmonary arterial hypertension may respond to therapy, evaluation for this should be considered in persistently dyspneic patients.
SKIN
Skin involvement is eventually identified in over a third of patients with sarcoidosis. The classic cutaneous lesions include erythema nodosum, maculopapular lesions, hyper- and hypopigmentation, keloid formation, and subcutaneous nodules. A specific complex of involvement of the bridge of the nose, the area beneath the eyes, and the cheeks is referred to as lupus pernio (Fig. 390-4) and is diagnostic for a chronic form of sarcoidosis.
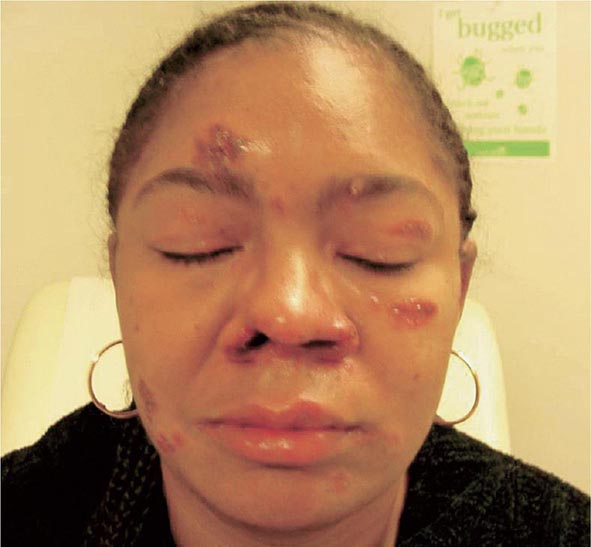
FIGURE 390-4 Chronic inflammatory lesions around nose, eyes, and cheeks, referred to as lupus pernio.
In contrast, erythema nodosum is a transient rash that can be seen in association with hilar adenopathy and uveitis (Löfgren’s syndrome). Erythema nodosum is more common in women and in certain self-described demographic groups including whites and Puerto Ricans. In the United States, the other manifestations of skin sarcoidosis, especially lupus pernio, are more common in African Americans than whites.
The maculopapular lesions from sarcoidosis are the most common chronic form of the disease (Fig. 390-5). These are often overlooked by the patient and physician, because they are chronic and not painful. Initially, these lesions are usually purplish papules and are often indurated. They can become confluent and infiltrate large areas of the skin. With treatment, the color and induration may fade. Because these lesions are caused by noncaseating granulomas, the diagnosis of sarcoidosis can be readily made by a skin biopsy.
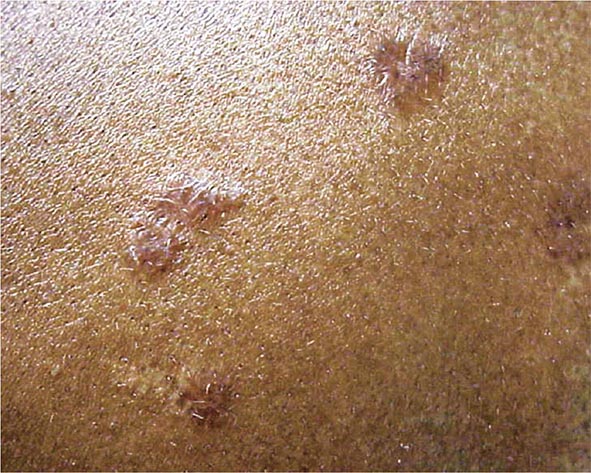
FIGURE 390-5 Maculopapular lesions on the trunk of a sarcoidosis patient.
EYE
The frequency of ocular manifestations for sarcoidosis varies depending on race. In Japan, >70% of sarcoidosis patients develop ocular disease, whereas in the United States only 30% have eye disease, with problems more common in African Americans than whites. Although the most common manifestation is an anterior uveitis, over a quarter of patients will have inflammation at the posterior of the eye, including retinitis and pars planitis. Although symptoms such as photophobia, blurred vision, and increased tearing can occur, some asymptomatic patients still have active inflammation. Initially asymptomatic patients with ocular sarcoidosis can eventually develop blindness. Therefore, it is recommended that all patients with sarcoidosis receive a dedicated ophthalmologic examination. Sicca is seen in over one-half of the chronic sarcoidosis patients. Dry eyes appear to be a reflection of prior lacrimal gland disease. Although the patient may no longer have active inflammation, the dry eyes may require natural tears or other lubricants.
LIVER
Using biopsies to detect granulomatous disease, liver involvement can be identified in over one-half of sarcoidosis patients. However, using liver function studies, only 20–30% of patients will have evidence of liver involvement. The most common abnormality of liver function is an elevation of the alkaline phosphatase level, consistent with an obstructive pattern. In addition, elevated transaminase levels can occur. An elevated bilirubin level is a marker for more advanced liver disease. Overall, only 5% of sarcoidosis patients have sufficient symptoms from their liver disease to require specific therapy. Although symptoms can be due to hepatomegaly, more frequently symptoms result from extensive intrahepatic cholestasis leading to portal hypertension. In this case, ascites and esophageal varices can occur. It is rare that a sarcoidosis patient will require a liver transplant, because even the patient with cirrhosis due to sarcoidosis can respond to systemic therapy. On a cautionary note, patients with both sarcoidosis and hepatitis C should avoid therapy with interferon a because of its association with the development or worsening of granulomatous disease.
BONE MARROW AND SPLEEN
One or more bone marrow manifestations can be identified in many sarcoidosis patients. The most common hematologic problem is lymphopenia, which is a reflection of sequestration of the lymphocytes into the areas of inflammation. Anemia occurs in 20% of patients, and leukopenia is less common. Bone marrow examination will reveal granulomas in about a third of patients. Although splenomegaly can be detected in 5–10% of patients, splenic biopsy reveals granulomas in 60% of patients. The CT scan can be relatively specific for sarcoidosis involvement of the spleen (Fig. 390-6). Both bone marrow and spleen involvement are more common in African Americans than whites. Although these manifestations alone are rarely an indication for therapy, on rare occasion, splenectomy may be indicated for massive symptomatic splenomegaly or profound pancytopenia. Nonthoracic lymphadenopathy can occur in up to 20% of patients.
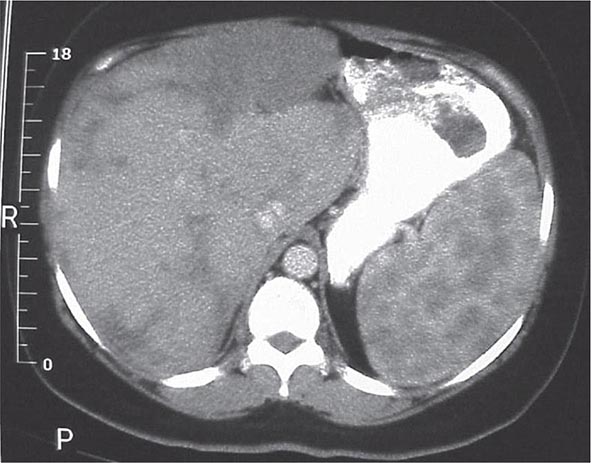
FIGURE 390-6 Computed tomography scan of the abdomen after oral and intravenous contrast. The stomach is compressed by the enlarged spleen. Within the spleen, areas of hypo- and hyperdensity are identified.
CALCIUM METABOLISM
Hypercalcemia and/or hypercalciuria occurs in about 10% of sarcoidosis patients. It is more common in whites than African Americans and in men. The mechanism of abnormal calcium metabolism is increased production of 1,25-dihydroxyvitamin D by the granuloma itself. The 1,25-dihydroxyvitamin D causes increased intestinal absorption of calcium, leading to hypercalcemia with a suppressed parathyroid hormone (PTH) level (Chap. 424). Increased exogenous vitamin D from diet or sunlight exposure may exacerbate this problem. Serum calcium should be determined as part of the initial evaluation of all sarcoidosis patients, and a repeat determination may be useful during the summer months with increased sun exposure. In patients with a history of renal calculi, a 24-h urine calcium measurement should be obtained. If a sarcoidosis patient with a history of renal calculi is to be placed on calcium supplements, a follow-up 24-h urine calcium level should be measured.
RENAL DISEASE
Direct kidney involvement occurs in <5% of sarcoidosis patients. It is associated with granulomas in the kidney itself and can lead to nephritis. However, hypercalcemia is the most likely cause of sarcoidosis-associated renal disease. In 1–2% of sarcoidosis patients, acute renal failure may develop as a result of hypercalcemia. Successful treatment of hypercalcemia with glucocorticoids and other therapies often improves but usually does not totally resolve the renal dysfunction.
NERVOUS SYSTEM
Neurologic disease is reported in 5–10% of sarcoidosis patients and appears to be of equal frequency across all ethnic groups. Any part of the central or peripheral nervous system can be affected. The presence of granulomatous inflammation is often visible on magnetic resonance imaging (MRI) studies. The MRI with gadolinium enhancement may demonstrate space-occupying lesions, but the MRI can be negative due to small lesions or the effect of systemic therapy in reducing the inflammation. The cerebral spinal fluid (CSF) findings include lymphocytic meningitis with a mild increase in protein. The CSF glucose is usually normal but can be low. Certain areas of the nervous system are more commonly affected in neurosarcoidosis. These include cranial nerve involvement, basilar meningitis, myelopathy, and anterior hypothalamic disease with associated diabetes insipidus (Chap. 404). Seizures and cognitive changes also occur. Of the cranial nerves, seventh nerve paralysis can be transient and mistaken for Bell’s palsy (idiopathic seventh nerve paralysis). Because this form of neurosarcoidosis often resolves within weeks and may not recur, it may have occurred prior to a definitive diagnosis of sarcoidosis. Optic neuritis is another cranial nerve manifestation of sarcoidosis. This manifestation is more chronic and usually requires long-term systemic therapy. It can be associated with both anterior and posterior uveitis. Differentiating between neurosarcoidosis and multiple sclerosis can be difficult at times. Optic neuritis can occur in both diseases. In some patients with sarcoidosis, multiple enhancing white matter abnormalities may be detected by MRI, suggesting multiple sclerosis. In such cases, the presence of meningeal enhancement or hypothalamic involvement suggests neurosarcoidosis, as does evidence of extraneurologic disease such as pulmonary or skin involvement, which also suggests sarcoidosis. Because the response of neurosarcoidosis to glucocorticoids and cytotoxic therapy is different from that of multiple sclerosis, differentiating between these disease entities is important.
CARDIAC
The presence of cardiac involvement is influenced by race. Although over a quarter of Japanese sarcoidosis patients develop cardiac disease, only 5% of sarcoidosis patients in the United States and Europe develop symptomatic cardiac disease. However, there is no apparent racial predilection between whites and African Americans. Cardiac disease, which usually presents as either congestive heart failure or cardiac arrhythmias, results from infiltration of the heart muscle by granulomas. Diffuse granulomatous involvement of the heart muscle can lead to profound dysfunction with left ventricular ejection fractions below 10%. Even in this situation, improvement in the ejection fraction can occur with systemic therapy. Arrhythmias can also occur with diffuse infiltration or with more patchy cardiac involvement. If the atrioventricular (AV) node is infiltrated, heart block can occur, which can be detected by routine electrocardiography. Ventricular arrhythmias and sudden death due to ventricular tachycardia are common causes of death. Arrhythmias are best detected using 24-h ambulatory monitoring, and electrophysiology studies may be negative. Other screening tests for cardiac disease include routine electrocardiography and echocardiography. The confirmation of cardiac sarcoidosis is usually performed with either MRI or positron emission tomography (PET) scanning. Because ventricular arrhythmias are usually multifocal due to patchy multiple granulomas in the heart, ablation therapy is not useful. Patients with significant ventricular arrhythmias should be considered for an implanted defibrillator, which appears to have reduced the rate of death in cardiac sarcoidosis. Although systemic therapy can be useful in treating the arrhythmias, patients may still have malignant arrhythmias up to 6 months after starting successful treatment, and the risk for recurrent arrhythmias occurs whenever medications are tapered.
MUSCULOSKELETAL SYSTEM
Direct granulomatous involvement of bone and muscle can be documented by radiography (x-ray, MRI, PET scan [Fig. 390-7], or gallium scan) or confirmed by biopsy in about 10% of sarcoidosis patients. However, a larger percentage of sarcoidosis patients complain of myalgias and arthralgias. These complaints are similar to those reported by patients with other inflammatory diseases, including chronic infections such as mononucleosis. Fatigue associated with sarcoidosis may be overwhelming for many patients. Recent studies have demonstrated a link between fatigue and small peripheral nerve fiber disease in sarcoidosis.
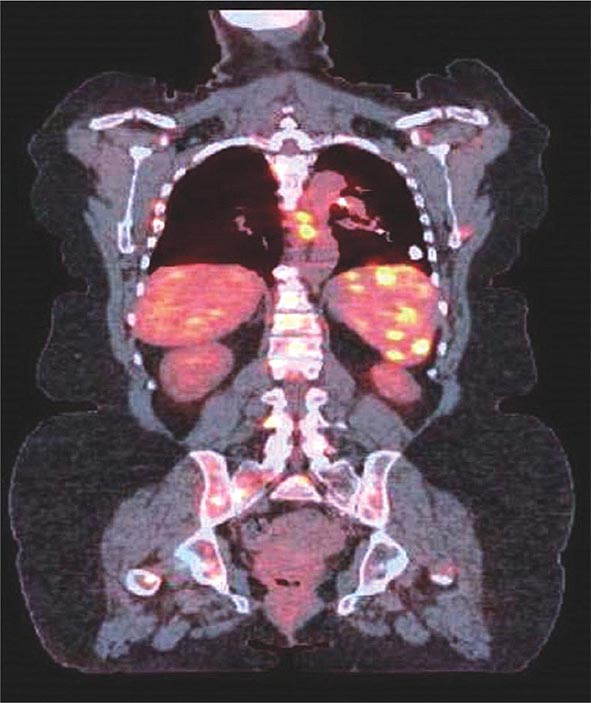
FIGURE 390-7 Positron emission tomography and computed tomography scan merged demonstrating increased activity in spleen, ribs, and spine of patient with sarcoidosis.
OTHER ORGAN INVOLVEMENT
Although sarcoidosis can affect any organ of the body, rarely does it involve the breast, testes, ovary, or stomach. Because of the rarity of involvement, a mass in one of these areas requires a biopsy to rule out other diseases including cancer. For example, in a study of breast problems in female sarcoidosis patients, a breast lesion was more likely to be a granuloma from sarcoidosis than from breast cancer. However, findings on the physical examination or mammogram cannot reliably differentiate between these lesions. More importantly, as women with sarcoidosis age, breast cancer becomes more common. Therefore, it is recommended that routine screening including mammography be performed along with other imaging studies (ultrasound, MRI) or biopsy as clinically indicated.
COMPLICATIONS
Sarcoidosis is usually a self-limited, non-life-threatening disease. However, organ-threatening disease can occur. These complications can include blindness, paraplegia, or renal failure. Death from sarcoidosis occurs in about 5% of patients seen in sarcoidosis referral clinics. The usual causes of death related to sarcoidosis are from lung, cardiac, neurologic, or liver involvement. In respiratory failure, an elevation of the right atrial pressure is a poor prognostic finding. Lung complications can also include infections such as mycetoma, which can subsequently lead to massive bleeding. In addition, the use of immunosuppressive agents can increase the incidence of serious infections.
LABORATORY FINDINGS
The chest roentgenogram remains the most commonly used tool to assess lung involvement in sarcoidosis. As noted above, the chest roentgenogram classifies involvement into four stages, with stages 1 and 2 having hilar and paratracheal adenopathy. The CT scan has been used increasingly in evaluating interstitial lung disease. In sarcoidosis, the presence of adenopathy and a nodular infiltrate is not specific for sarcoidosis. Adenopathy up to 2 cm can be seen in other inflammatory lung diseases such as idiopathic pulmonary fibrosis. However, adenopathy >2 cm in the short axis supports the diagnosis of sarcoidosis over other interstitial lung diseases.
The PET scan has increasingly replaced gallium-67 scanning to identify areas of granulomatous disease in the chest and other parts of the body (Fig. 390-7). Both tests can be used to identify potential areas for biopsy. Cardiac PET scanning has also proved useful in assessing cardiac sarcoidosis. The identification of hypermetabolic activity may be due to the granulomas from sarcoidosis and not to disseminated malignancy.
MRI has also proved useful in the assessment of extrapulmonary sarcoidosis. Gadolinium enhancement has been demonstrated in areas of inflammation in the brain, heart, and bone. MRI scans may detect asymptomatic lesions. Like PET scan, MRI changes appear similar to those seen with malignancy and infection. In some cases, biopsy may be necessary to determine the cause of the radiologic abnormality.
Serum levels of angiotensin-converting enzyme (ACE) can be helpful in the diagnosis of sarcoidosis. However, the test has somewhat low sensitivity and specificity. Elevated levels of ACE are reported in 60% of patients with acute disease and only 20% of patients with chronic disease. Although there are several causes for mild elevation of ACE, including diabetes, elevations of >50% of the upper limit of normal are seen in only a few conditions including sarcoidosis, leprosy, Gaucher’s disease, hyperthyroidism, and disseminated granulomatous infections such as miliary tuberculosis. Because the ACE level is determined by a biologic assay, the concurrent use of an ACE inhibitor such as lisinopril will lead to a very low ACE level.
DIAGNOSIS
The diagnosis of sarcoidosis requires both compatible clinical features and pathologic findings. Because the cause of sarcoidosis remains elusive, the diagnosis cannot be made with 100% certainty. Nevertheless, the diagnosis can be made with reasonable certainty based on history and physical features along with laboratory and pathologic findings.
Patients are usually evaluated for possible sarcoidosis based on two scenarios (Fig. 390-8). In the first scenario, a patient may undergo a biopsy revealing a noncaseating granuloma in either a pulmonary or an extrapulmonary organ. If the clinical presentation is consistent with sarcoidosis and there is no alternative cause for the granulomas identified, then the patient is felt to have sarcoidosis.
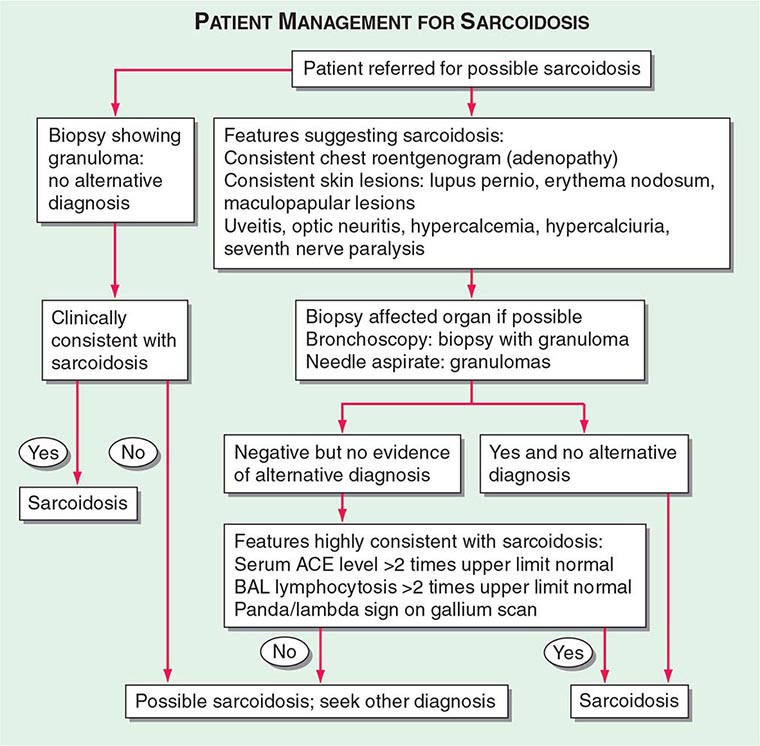
FIGURE 390-8 Proposed approach to management of patient with possible sarcoidosis. Presence of one or more of these features supports the diagnosis of sarcoidosis: uveitis, optic neuritis, hypercalcemia, hypercalciuria, seventh cranial nerve paralysis, diabetes insipidus. ACE, angiotensin-converting enzyme; BAL, bronchoalveolar lavage.
In the second scenario, signs or symptoms suggesting sarcoidosis such as the presence of bilateral adenopathy may be present in an otherwise asymptomatic patient or a patient with uveitis or a rash consistent with sarcoidosis. At this point, a diagnostic procedure should be performed. For the patient with a compatible skin lesion, a skin biopsy should be considered. Other biopsies to consider could include liver, extrathoracic lymph node, or muscle. In some cases, a biopsy of the affected organ may not be easy to perform (such as a brain or spinal cord lesion). In other cases, such as an endomyocardial biopsy, the likelihood of a positive biopsy is low. Because of the high rate of pulmonary involvement in these cases, the lung may be easier to approach by bronchoscopy. During the bronchoscopy, a transbronchial biopsy, bronchial biopsy, or transbronchial needle aspirate can be performed. The endobronchial ultrasonography-guided (EBUS) transbronchial needle aspirate can assist in diagnosing sarcoidosis in patients with mediastinal adenopathy (stage 1 or 2 radiographic pulmonary disease), whereas transbronchial biopsy has a higher diagnostic yield for those with only parenchymal lung disease (stage 3). These tests are complementary and may be performed together.
If the biopsy reveals granulomas, an alternative diagnosis such as infection or malignancy must be excluded. Bronchoscopic washings can be sent for cultures for fungi and tuberculosis. For the pathologist, the more tissue that is provided, the more comfortable is the diagnosis of sarcoidosis. A needle aspirate may be adequate in an otherwise classic case of sarcoidosis, but may be insufficient in a patient in whom lymphoma or fungal infection is a likely alternative diagnosis. Because granulomas can be seen on the edge of a lymphoma, the presence of a few granulomas from a needle aspirate may not be sufficient to clarify the diagnosis. Mediastinoscopy provides a larger sample to confirm the presence or absence of lymphoma in the mediastinum. Alternatively, for most patients, evidence of extrathoracic disease (e.g., eye involvement) may further support the diagnosis of sarcoidosis.
For patients with negative pathology, positive supportive tests may increase the likelihood of the diagnosis of sarcoidosis. These tests include an elevated ACE level, which can also be elevated in other granulomatous diseases but not in malignancy. A positive PET scan can support the diagnosis if multiple organs are affected. A BAL is often performed during the bronchoscopy. An increase in the percentage of lymphocytes supports the diagnosis of sarcoidosis. The use of the lymphocyte markers CD4 and CD8 can be used to determine the CD4/CD8 ratio of these increased lymphocytes in the BAL fluid. A ratio of >3.5 is strongly supportive of sarcoidosis but is less sensitive than an increase in lymphocytes alone. Although in general, an increase in BAL lymphocytes is supportive of the diagnosis, other conditions must be considered.
Supportive findings, when combined with commonly associated but nondiagnostic clinical features of the disease, improve the diagnostic probability of sarcoidosis. These clinical features include uveitis, renal stones, hypercalcemia, seventh cranial nerve paralysis, or erythema nodosum. The presence of one or more of these features in a patient suspected of having sarcoidosis increases the probability of sarcoidosis.
The Kviem-Siltzbach procedure is a specific diagnostic test for sarcoidosis. An intradermal injection of specially prepared tissue derived from the spleen of a known sarcoidosis patient is biopsied 4–6 weeks after injection. If noncaseating granulomas are seen, this is highly specific for the diagnosis of sarcoidosis. Unfortunately, there is no commercially available Kviem-Siltzbach reagent, and some locally prepared batches have lower specificity. Thus, this test is of historic interest and is rarely used in current clinical practice.
Because the diagnosis of sarcoidosis can never be certain, over time other features may arise that lead to an alternative diagnosis. Conversely, evidence for new organ involvement may eventually confirm the diagnosis of sarcoidosis.
PROGNOSIS
The risk of death or loss of organ function remains low in sarcoidosis. Poor outcomes usually occur in patients who present with advanced disease in whom treatment seems to have little impact. In these cases, irreversible fibrotic changes have frequently occurred. Over the past 20 years, the reported mortality from sarcoidosis has increased in the United States and England. Whether this is due to heightened awareness of the chronic nature of this disease or to other factors such as more widespread immunosuppressive therapy usage remains unclear.
For the majority of patients, initial presentation occurs during the granulomatous phase of the disease as depicted in Fig. 390-1. It is clear that many patients resolve their disease within 2–5 years. These patients are felt to have acute, self-limiting sarcoidosis. However, there is a form of the disease that does not resolve within the first 2–5 years. These chronic patients can be identified at presentation by certain risk factors at presentation such as fibrosis on chest roentgenogram, presence of lupus pernio, bone cysts, cardiac or neurologic disease (except isolated seventh nerve paralysis), and presence of renal calculi due to hypercalciuria. Recent studies also indicate that patients who require glucocorticoids for any manifestation of their disease in the first 6 months of presentation have a >50% chance of having chronic disease. In contrast, <10% of patients who require no systemic therapy in the first 6 months will require chronic therapy.
TREATMENT | SARCOIDOSIS |
Indications for therapy should be based on symptoms or presence of organ- or life-threatening disease, including disease involving the eye, heart, or nervous system. The patient with asymptomatic elevated liver function tests or an abnormal chest roentgenogram probably does not benefit from treatment. However, these patients should be monitored for evidence of progressive, symptomatic disease.
One approach to therapy is summarized in Figs. 390-9 and 390-10. We have divided the approach into treating acute versus chronic disease. For acute disease, no therapy remains a viable option for patients with no or mild symptoms. For symptoms confined to only one organ, topical therapy is preferable. For multiorgan disease or disease too extensive for topical therapy, an approach to systemic therapy is outlined. Glucocorticoids remain the drugs of choice for this disease. However, the decision to continue to treat with glucocorticoids or to add steroid-sparing agents depends on the tolerability, duration, and dosage of glucocorticoids.Table 390-2 summarizes the dosage and monitoring of several commonly used drugs. According to the available trials, evidence-based recommendations are made. Most of these recommendations are for pulmonary disease because most of the trials were performed only in pulmonary disease. Treatment recommendations for extrapulmonary disease are usually similar with a few modifications. For example, the dosage of glucocorticoids is usually higher for neurosarcoidosis and lower for cutaneous disease. There was some suggestion that higher doses would be beneficial for cardiac sarcoidosis, but one study found that initial prednisone doses >40 mg/d were associated with a worse outcome because of toxicity.
COMMONLY USED DRUGS TO TREAT SARCOIDOSIS |
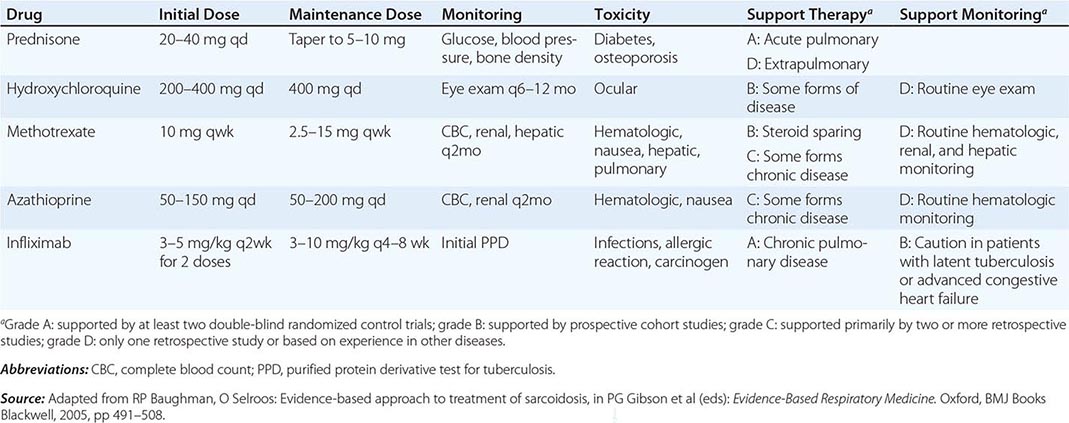
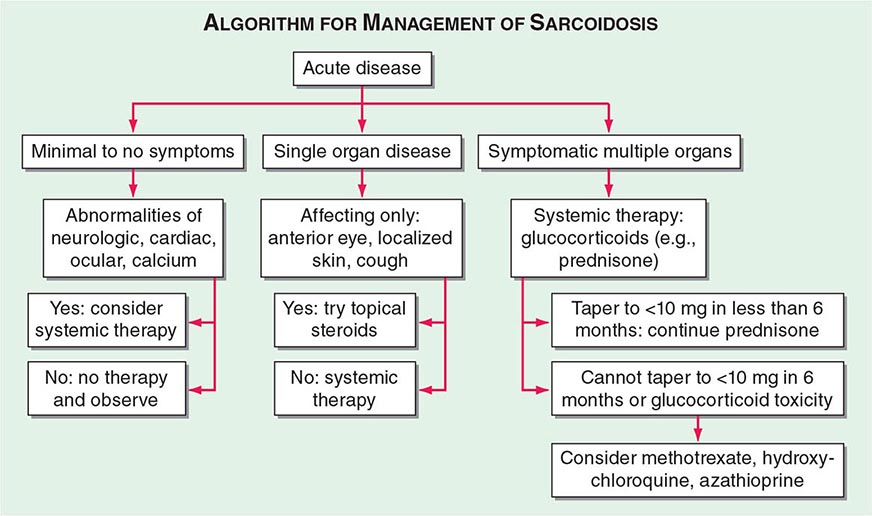
FIGURE 390-9 The management of acute sarcoidosis is based on level of symptoms and extent of organ involvement. In patients with mild symptoms, no therapy may be needed unless specified manifestations are noted.
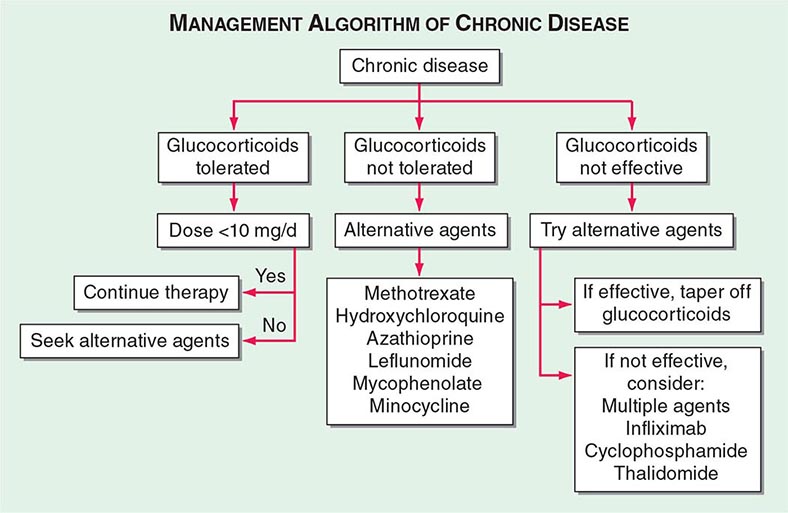
FIGURE 390-10 Approach to chronic disease is based on whether glucocorticoid therapy is tolerated or not.
Systemic therapies for sarcoidosis are usually immunosuppressive including glucocorticoids, cytotoxics, or biologics. Although most patients receive glucocorticoids as their initial systemic therapy, toxicity associated with prolonged therapy often leads to steroid-sparing alternatives. The antimalarial drugs such as hydroxychloroquine are more effective for skin than pulmonary disease. Minocycline may also be useful for cutaneous sarcoidosis. For pulmonary and other extrapulmonary disease, cytotoxic agents are often used. These include methotrexate, azathioprine, leflunomide, mycophenolate, and cyclophosphamide. The most widely studied cytotoxic agent has been methotrexate. This agent works in approximately two-thirds of sarcoidosis patients, regardless of the disease manifestation. In one retrospective study comparing methotrexate to azathioprine, both drugs were equally effective. However, methotrexate was associated with significantly less toxicity. As noted in Table 390-2, specific guidelines for monitoring therapy have been recommended. Cytokine modulators such as thalidomide and pentoxifylline have also been used in a limited number of cases.
The biologic anti-TNF agents have recently been studied in sarcoidosis, with prospective randomized trials completed for both etanercept and infliximab. Etanercept has a limited role as a steroid-sparing agent. Conversely, infliximab significantly improved lung function when administered to glucocorticoid and cytotoxic pretreated patients with chronic disease The difference in response between these two agents is similar to that observed in Crohn’s disease, where infliximab is effective and etanercept is not. However, there is a higher risk for reactivation of tuberculosis with infliximab compared to etanercept. The differential response rate could be explained by differences in mechanism of action because etanercept is a TNF receptor antagonist and infliximab is a monoclonal antibody against TNF. In contrast to etanercept, infliximab also binds to TNF on the surface of some cells that release TNF, which leads to cell lysis. This effect has been documented in Crohn’s disease. Adalimumab is a humanized monoclonal anti-TNF antibody that also appears effective for sarcoidosis when dosed at higher strengths, as recommended for the treatment of Crohn’s disease. The role of the newer therapeutic agents for sarcoidosis is still evolving. However, these targeted therapies confirm that TNF may be an important target, especially in the treatment of chronic disease. However, these agents are not a panacea, because sarcoidosis-like disease has occurred in patients treated with anti-TNF agents for nonsarcoidosis indications.
391e | IgG4-Related Disease |
IgG4-related disease (IgG4-RD) is a fibroinflammatory condition characterized by a tendency to form tumefactive lesions. The clinical manifestations of this disease, however, are protean, and continue to be defined. IgG4-RD has now been described in virtually every organ system. Commonly affected organs are the biliary tree, salivary glands, periorbital tissues, kidneys, lungs, lymph nodes, and retroperitoneum. In addition, IgG4-RD involvement of the meninges, aorta, prostate, thyroid, pericardium, skin, and other organs is well described. The disease is believed to affect the brain parenchyma, the joints, the bone marrow, and the bowel mucosa only rarely (if ever).
The clinical features of IgG4-RD are numerous, but the pathologic findings are consistent across all affected organs. These findings include a lymphoplasmacytic infiltrate with a high percentage of IgG4-positive plasma cells; a characteristic pattern of fibrosis termed “storiform”; a tendency to target blood vessels, particularly veins, through an obliterative process (“obliterative phlebitis”); and a mild to moderate tissue eosinophilia.
IgG4-RD encompasses a number of conditions previously regarded as separate, organ-specific entities. A condition once known as “lymphoplasmacytic sclerosing pancreatitis” (among many other terms) became the paradigm of IgG4-RD in 2000, when Japanese investigators recognized that these patients had elevated serum concentrations of IgG4. This form of sclerosing pancreatitis is now termed type 1 (IgG4-related) autoimmune pancreatitis (AIP). By 2003, extrapancreatic disease manifestations had been identified in patients with type 1 AIP, and since then, the manifestations of IgG4-RD in many organs have been catalogued. Mikulicz’s disease, once considered to be a subset of Sjögren’s syndrome that affected the lacrimal, parotid, and submandibular glands, is now considered part of the IgG4-RD spectrum. Similarly, a subset of patients previously diagnosed as having primary sclerosing cholangitis was known to respond well to glucocorticoids, in contrast to the majority of patients with that diagnosis. This steroid-responsive subset is now explained by the fact that such patients actually have a separate disease, i.e., IgG4-related sclerosing cholangitis. In this manner, the understanding of IgG4-RD has extended to include nearly every specialty of medicine.
CLINICAL FEATURES
The major organ lesions are summarized in Table 391e-1. IgG4-RD usually presents subacutely, and most patients do not have severe constitutional symptoms. Fevers and dramatic elevations of C-reactive protein are unusual; however, some patients report substantial weight loss occurring over periods of months. Clinically apparent disease can evolve over months, years, or even decades before the manifestations within a given organ becomes sufficiently severe to bring the patient to medical attention. Some patients have disease that is marked by the appearance and then resolution or temporary improvement in symptoms within a particular organ. Other patients accumulate new organ involvement as their disease persists in previously affected organs. Many patients with IgG4-RD are misdiagnosed as having other conditions, particularly malignancies, or their findings are attributed initially to nonspecific inflammation. The disorder is often identified incidentally through radiologic findings or unexpectedly in pathology specimens.
ORGAN MANIFESTATIONS OF IGG4-RELATED DISEASE |
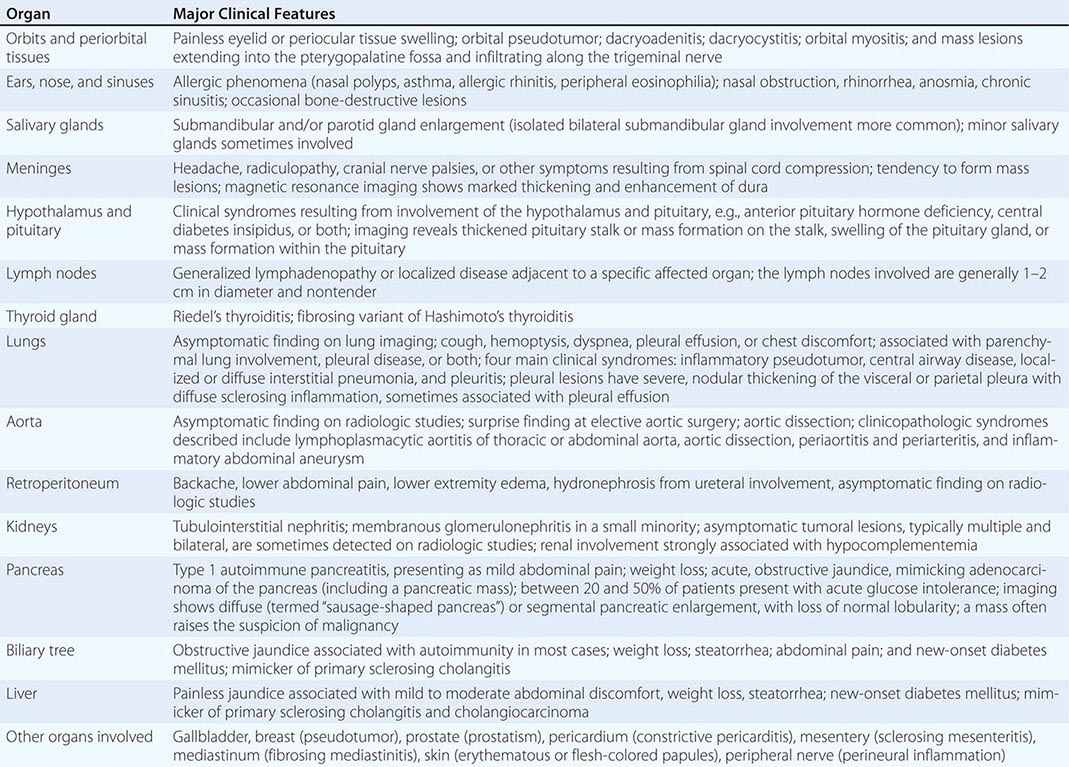
Multiorgan disease may be evident at diagnosis but can also evolve over months to years. Some patients have disease confined to a single organ for many years. Others have either known or subclinical organ involvement at the same time as the major clinical feature. Patients with type 1 AIP may have their major disease focus in the pancreas; however, thorough evaluations by history, physical examination, blood tests, urinalysis, and cross-sectional imaging may demonstrate lacrimal gland enlargement, sialoadenitis, lymphadenopathy, a variety of pulmonary findings, tubulointerstitial nephritis, hepatobiliary disease, aortitis, retroperitoneal fibrosis, or other organ involvement. Spontaneous improvement, sometimes leading to clinical resolution of certain organ system manifestations, is reported in a small percentage of patients.
Two common characteristics of IgG4-RD are allergic disease and the tendency to form tumefactive lesions that mimic malignancies (Fig. 391e-1). Many IgG4-RD patients have allergic features such as atopy, eczema, asthma, nasal polyps, sinusitis, and modest peripheral eosinophilia. IgG4-RD also appears to account for a significant proportion of tumorous swellings—pseudotumors—in many organ systems. Some patients undergo major surgeries (e.g., Whipple procedures or thyroidectomy) for the purpose of resecting malignancies before the correct diagnosis is identified. Frequent sites of pseudotumors are the major salivary glands, lacrimal glands, lungs, and kidneys; however, nearly all organs have been affected with this manifestation.
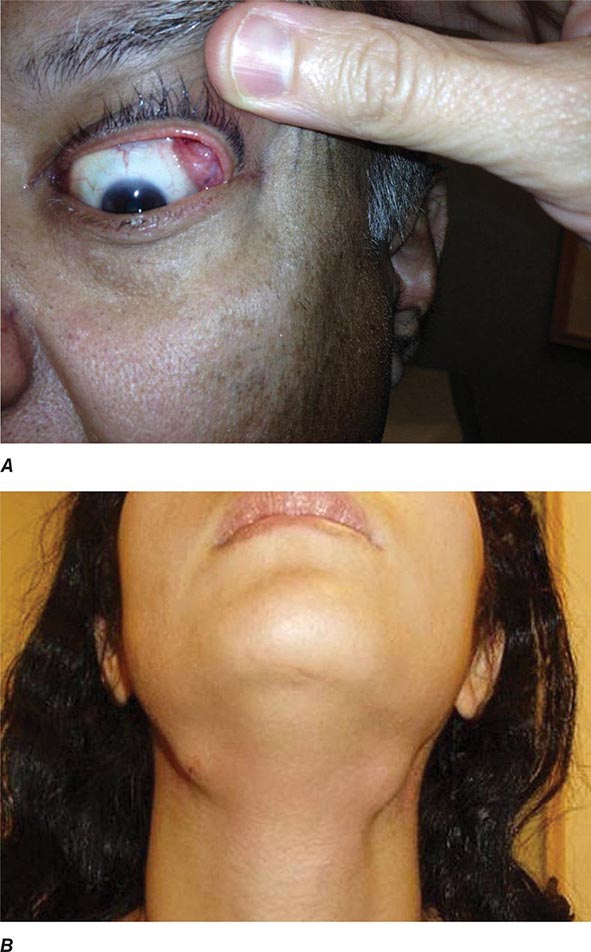
FIGURE 391e-1 A major clinical feature of IgG4-related disease is its tendency to form tumefactive lesions. Shown here are mass lesions of the lacrimal glands (A) and the submandibular glands (B).
IgG4-RD often causes major morbidity and can lead to organ failure; however, its general pattern is to cause damage in a subacute manner. Destructive bone lesions in the sinuses, head, and middle ear spaces that mimic granulomatous polyangiitis (formerly Wegener’s granulomatosis) also occur in IgG4-RD; less aggressive lesions are the rule in most organs. In regions such as the retroperitoneum, substantial fibrosis often occurs before the diagnosis is established, leading to ureteral entrapment, hydronephrosis, postobstructive uropathy, renal atrophy, and chronic pain, possibly resulting from the encasement of peripheral nerves by the inflammatory process. Undiagnosed or undertreated IgG4-related cholangitis can lead to hepatic failure within months. Similarly, IgG4-related aortitis, believed to be associated with between 10 and 50% of cases of inflammatory aortitis, can cause aneurysms and dissections. Substantial renal dysfunction and even renal failure can ensue from IgG4-related tubulointerstitial nephritis, and renal atrophy is a frequent sequel to this disease complication.
SEROLOGIC FINDINGS
The majority of patients with IgG4-RD have elevated serum IgG4 concentrations; however, the range of elevation varies widely. Serum concentrations of IgG4 as high as 30 or 40 times the upper limit of normal sometimes occur, usually in patients with disease that affects multiple organ systems simultaneously. Approximately 30% of patients have normal serum IgG4 concentrations despite classic histopathologic and immunohistochemical findings. Such patients tend to have disease that affects fewer organs. Patients with IgG4-related retroperitoneal fibrosis have a high likelihood of normal serum IgG4 concentrations, perhaps because the process has advanced to a fibrotic stage by the time the diagnosis is considered.
The correlation between serum IgG4 concentrations and disease activity and the need for treatment is imperfect. Serum IgG4 concentrations typically decline swiftly with the institution of therapy but often do not normalize completely. Patients can achieve clinical remissions yet have persistently elevated serum IgG4 concentrations. Rapidly rising serum IgG4 concentrations may identify patients at greatest risk for clinical flares and monitoring of serial IgG4 concentrations identifies early relapse in some patients; however, the temporal relationship between modest IgG4 elevations and the need for clinical treatment is poor. Clinical relapses occur in some patients despite persistently normal IgG4 concentrations.
IgG4 concentrations in serum are usually measured by nephelometry assays. These assays can lead to reports of spuriously low IgG4 values because of the prozone effect. This effect can be corrected by dilution of the serum sample in the laboratory. The prozone effect should be considered when the results of serologic testing for IgG4 concentrations appear to be at odds with the patient’s clinical features.
EPIDEMIOLOGY
The typical patient with IgG4-RD is a middle-aged to elderly man. This epidemiology stands in stark contrast to that of many classic autoimmune conditions, which tend to affect young women. Studies of AIP patients in Japan indicate that the male-to-female ratio in that disease subset is on the order of 3:1. Even more striking, male predominance has been reported in IgG4-related tubulointerstitial nephritis and IgG4-related retroperitoneal fibrosis. Among IgG4-RD manifestations that involve organs of the head and neck, the sex ratio may be closer to 1:1.
PATHOLOGY
The key histopathology characteristics of IgG4-RD are a dense lymphoplasmacytic infiltrate (Fig. 391e-2) that is organized in a storiform pattern (resembling a basket-weave), obliterative phlebitis, and a mild to moderate eosinophilic infiltrate. Lymphoid follicles and germinal centers are frequently observed. The infiltrate tends to aggregate around ductal structures when it affects glands such as the lacrimal, submandibular, and parotid glands or the pancreas. The inflammatory lesion often aggregates into tumefactive masses that destroy the involved tissue.
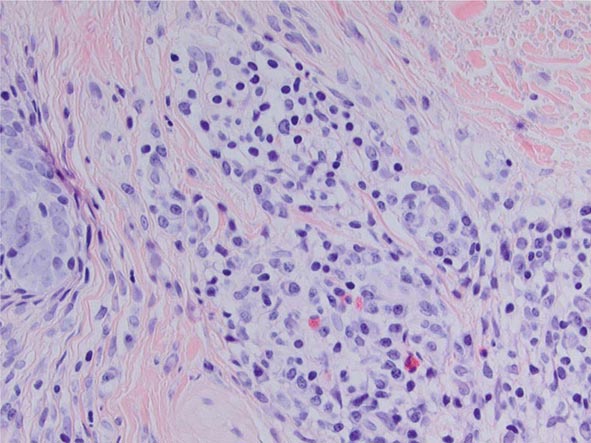
FIGURE 391e-2 Hallmark histopathology characteristics of IgG4-related disease (IgG4-RD) are a dense lymphoplasmacytic infiltrate and a mild to moderate eosinophilic infiltrate. The cellular inflammation is often encased in a distinctive type of fibrosis termed “storiform,” which often has a basket weave pattern. Abundant fibroblasts and strands of fibrosis accompany the lymphoplasmacytic infiltrate and eosinophils in this figure. This biopsy was taken from a nodular lesion on the cheek; however, the findings are identical to the pathology found in the pancreas, kidneys, lungs, salivary glands, and other organs affected by IgG4-RD.
Obliterative arteritis is observed in some organs, particularly the lung; however, venous involvement is more common (and is indeed a hallmark of IgG4-RD). Several histopathology features are uncommon in IgG4-RD and, when detected, mitigate against the diagnosis of IgG4-RD. These include intense neutrophilic infiltration, leukocytoclasis, granulomatous inflammation, multinucleated giant cells, and fibrinoid necrosis.
The inflammatory infiltrate is composed of an admixture of B and T lymphocytes. B cells are typically organized in germinal centers. Plasma cells staining for CD19, CD138, and IgG4 appear to radiate out from the germinal centers. In contrast, the T cells, usually CD4+, are distributed more diffusely throughout the lesion and generally represent the most abundant cell type. Fibroblasts, histiocytes, and eosinophils can all be observed in moderate numbers. Some biopsy samples are particularly enriched with eosinophils. In other samples, particularly from long-standing cases, fibrosis predominates.
The histologic appearance of IgG4-RD, although highly characteristic, requires immunohistochemical confirmation of the diagnosis with IgG4 immunostaining. IgG4-positive plasma cells predominate within the lesion, but plasma cells containing immunoglobulins from each subclass can be found. The number of IgG4-positive plasma cells can be quantified by either counting the number of cells per high-power field (HPF) or by calculating the ratio of IgG4- to IgG-bearing plasma cells. Tissue fibrosis predominates in the latter phases of organ involvement, and in this relatively acellular phase of inflammation, both the IgG4:total IgG ratio and the pattern of tissue fibrosis are more important than the number of IgG4-positive cells per HPF in establishing the diagnosis. In situ hybridization techniques are also now used to circumvent problems posed by increased background staining in conventional immunostaining techniques.
PATHOPHYSIOLOGY
The IgG4 molecule is believed to play an indirect role in the pathophysiology of disease in most organs. However, the molecule has properties that are unique among the immunoglobulin subclasses and that may contribute to tissue injury in some circumstances. As an example, IgG4 molecules have the ability to undergo Fab exchange, a phenomenon in which the two halves of the molecule dissociate from each other and reassociate with dissimilar hemi-molecules from other IgG4 molecules. This property is unique among the immunoglobulin subclasses. Partly as a result of Fab exchange, however, IgG4 antibodies bind antigen loosely. The molecules have low affinities for Fc receptors and C1q and are regarded generally as noninflammatory immunoglobulins. The low affinities for Fc receptors and C1q impair the ability of IgG4 antibodies to induce phagocyte activation, antibody-dependent cellular cytotoxicity, and complement-mediated damage. It is possible that the increased concentrations of IgG4 in serum and IgG4-bearing plasma cells in tissue are merely the result of other effector pathways, such as TH2/Treg cytokines, that are more central to the inflammation and tissue damage.
TREATMENT
Not every disease manifestation of IgG4-RD requires immediate treatment because the disease takes an indolent form in many patients. IgG4-related lymphadenopathy, for example, can be asymptomatic for years, without evolution to other disease manifestations. Thus, watchful waiting is prudent in some cases. Vital organ involvement must be treated aggressively, however, because IgG4-RD can lead to serious organ dysfunction and failure. Aggressive disease can lead quickly to end-stage liver disease, permanent impairment of pancreatic function, renal atrophy, aortic dissection or aneurysms, and destructive lesions in the sinuses and nasopharynx.
Glucocorticoids are the first line of therapy. Treatment regimens, extrapolated from experience with the management of type 1 AIP, generally begin with 40 mg/d of prednisone, with tapering to discontinuation or maintenance doses of 5 mg/d within 2 or 3 months. The clinical response to glucocorticoids is usually swift and striking; however, longitudinal data indicate that disease flares occur in more than 90% of patients within 3 years. Conventional steroid-sparing agents such as azathioprine and mycophenolate mofetil have been used in some patients; however, evidence for their efficacy is lacking.
For patients with relapsing or glucocorticoid-resistant disease, B cell depletion with rituximab is an excellent second-line therapy. Rituximab treatment (two doses of 1 g IV, separated by approximately 15 days) leads to a targeted, precipitous decline in serum IgG4 concentrations, suggesting that rituximab achieves its effects in part by preventing the repletion of short-lived plasma cells that produce IgG4. More important than its effects on IgG4 concentrations, however, may be the effect of B cell depletion on T cell function. Specific effects of rituximab on CD4+ effector T cells have been documented in IgG4-RD.
Rituximab may be an appropriate first-line therapy for some patients, particularly those at high risk for glucocorticoid toxicity and patients with immediately organ-threatening disease. The optimal approaches to remission maintenance, by either re-treatment with rituximab or continuous low-dose glucocorticoid therapy, require further study.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


