Chapter 17 Idiopathic connective tissue disorders
Lupus erythematosus
Lupus erythematosus is a complex disorder associated with numerous clinical signs and symptoms and a wide range of laboratory abnormalities. It shows a spectrum of varying prognosis, ranging from a benign, solely cutaneous variant (localized discoid) through to a potentially fatal systemic illness.1–4 The range of subtypes is shown in Table 17.1.
Table 17.1 Lupus erythematosus: subtypes
Although the precise etiology is unknown, it is thought that interplay of genetic factors, autoantibodies, immune complexes, hormones, and other factors is responsible for the development of the illness. There is evidence suggesting that the incidence of the systemic variant is increasing, but due to earlier diagnosis and more effective therapy, the mortality rate has significantly diminished and the 10-year overall survival rate in adults now exceeds 90%.5 The presence of renal and/or neurological involvement, however, remains a poor prognostic indicator.6
Pediatric systemic lupus erythematosus (SLE) is an aggressive illness with considerable mortality, largely due to the incidence of renal disease. Even with corticosteroid and immunosuppressive therapy the death rate is as high as 15%.7
Clinical features
Discoid lupus erythematosus
Discoid lupus erythematosus (DLE), the commonest form, is subdivided into localized and generalized variants. This is of prognostic importance because only about 1% of patients with localized DLE develop systemic disease, but approximately 5% of those with the generalized form (in particular those with persistent anemia, leukopenia, thrombocytopenia, false-positive Wassermann reaction, and high-titer antinuclear factor) develop full-blown SLE.6,8–10 Discoid lesions develop in up to 20% of patients with systemic lupus.11 Periungual telangiectasia, sclerodactyly, and the presence of Raynaud’s phenomenon may also signify potential disease progression.10 Patients with DLE should not be made unduly worried about the risk of developing systemic involvement, which is not high.
Discoid lupus erythematosus is persistent and affects twice as many females as males. Although any age group may be involved, it is most common in the third, fourth, and fifth decades, with a peak incidence in the late thirties.12 Presentation in childhood is rare.13–18 Lesions typically arise on sun-exposed sites and patients frequently experience photosensitivity; there may be spring and summer exacerbation.5 In the localized form, the head and neck are usually affected, but in the generalized variant, lesions may also be present on the dorsal aspect of the arms, hands, and fingers and on the ‘V’ of the neck. Nonexposed sites, including the trunk, upper limbs, and the palms and soles, are also commonly involved.19
Facial plaques occur most often on the cheeks (Fig. 17.1). Other sites affected include the bridge of the nose, the ears, the neck, and the scalp (Figs 17.2–17.6). The associated scarring of the scalp is followed by permanent alopecia (Figs 17.7,17.8). Scalp involvement, which is more common in those patients affected by the disease when they were young, is chronic, and correlates with longstanding severe illness.20
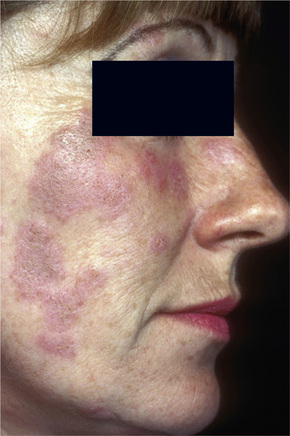
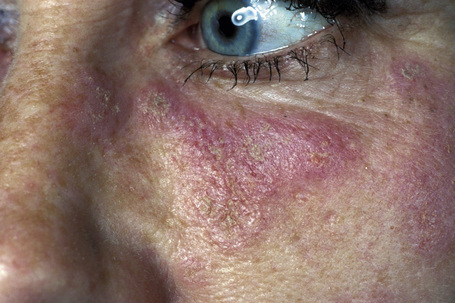
Fig. 17.2 Discoid lupus erythematosus: close-up view showing erythema and scale.
From the collection of the late N.P. Smith, MD, the Institute of Dermatology, London, UK.
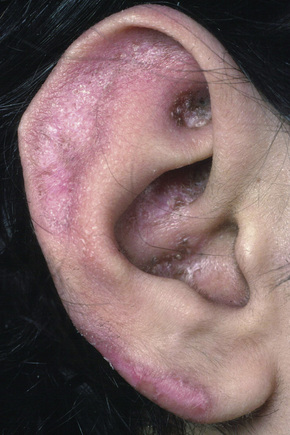
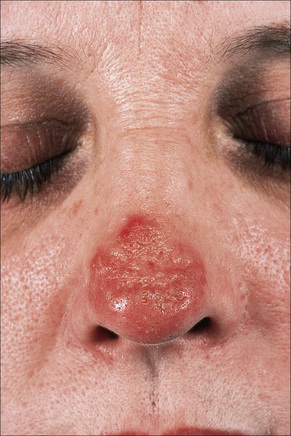
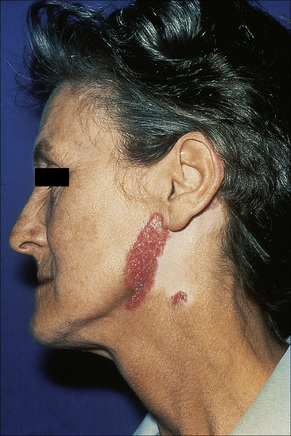
Fig. 17.5 Discoid lupus erythematosus: in this chronic lesion there is marked scarring.
By courtesy of R.A. Marsden, MD, St George’s Hospital, London, UK.
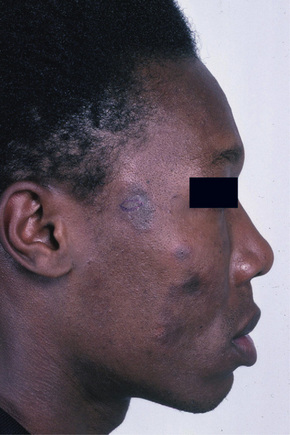
Fig. 17.6 Discoid lupus erythematosus: dark-skinned races are commonly affected.
From the collection of the late N.P. Smith, MD, the Institute of Dermatology, London, UK.
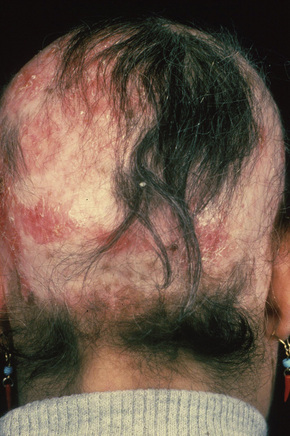
Fig. 17.7 Discoid lupus erythematosus: hair loss is permanent.
By courtesy of the Institute of Dermatology, London, UK.
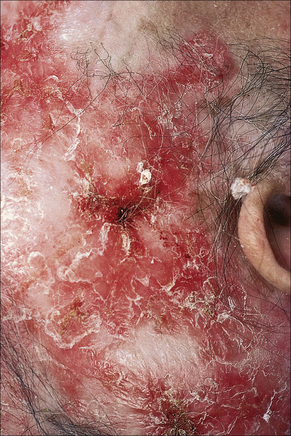
Fig. 17.8 Discoid lupus erythematosus: there is alopecia with very marked scarring.
By courtesy of the Institute of Dermatology, London, UK.
Early lesions appear edematous and erythematous. An established plaque of DLE, which may measure up to 10.0 cm across, is usually covered with an adherent scale accompanied by epidermal atrophy, follicular dilatation, and plugging (Figs 17.9, 17.10).5,19 If the scale is removed the horny plugs are often seen attached to its undersurface (‘carpet tacks’ sign). Telangiectasia is a common finding and lesions heal with scarring, which is often marked. In dark- or black-skinned individuals the plaque may be hypo- or hyperpigmented and this may be particularly disfiguring (Figs 17.11–17.14).19
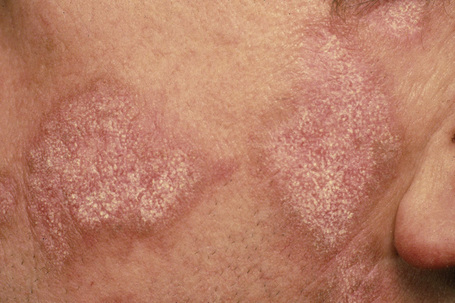
Fig. 17.9 Discoid lupus erythematosus: close-up view of scale. Note the erythematous border.
By courtesy of the Institute of Dermatology, London, UK.
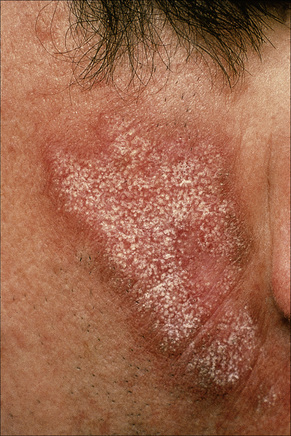
Fig. 17.10 Discoid lupus erythematosus: close-up view showing follicular plugging.
By courtesy of the Institute of Dermatology, London, UK.
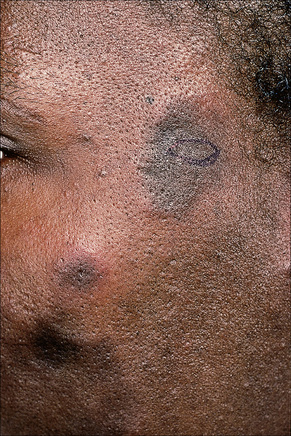
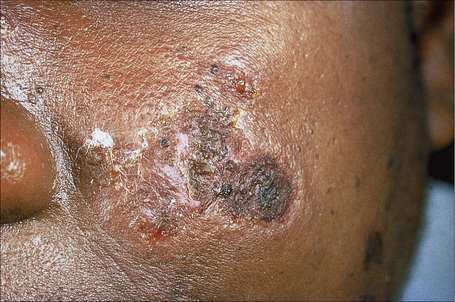
Fig. 17.12 Discoid lupus erythematosus: close-up view.
By courtesy of R.A. Marsden, MD, St George’s Hospital, London, UK.
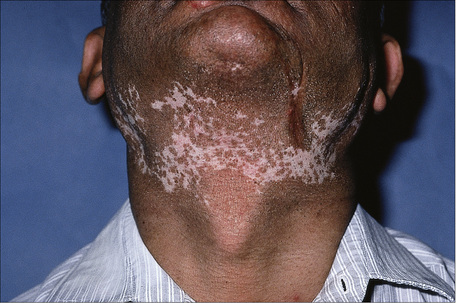
Fig. 17.13 Discoid lupus erythematosus: marked hypopigmentation may be a distressing complication.
By courtesy of R.A. Marsden, MD, St George’s Hospital, London, UK.
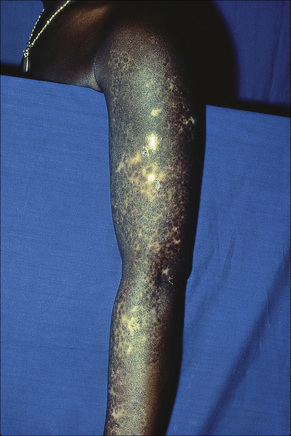
Fig. 17.14 Discoid lupus erythematosus: in this patient, the arm is severely affected.
By courtesy of R.A. Marsden, MD, St George’s Hospital, London, UK.
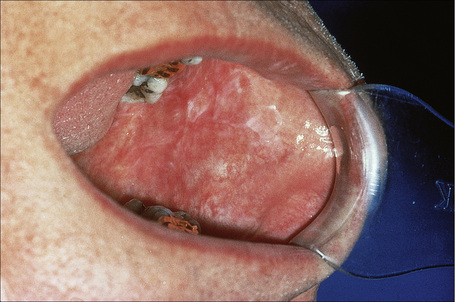
Oral involvement occurs in 20–25% of patients with DLE, with the vermilion border of the lower lip, alveolar processes, labial and buccal mucosae being particularly affected (Figs 17.15–17.17).21–26 Chronic lesions are typically erythematous and atrophic with a scalloped white keratotic border and adjacent telangiectasia.22 Erosions and ulcers are additional features.12 Lesions are sometimes indistinguishable from atrophic lichen planus. Involvement of the tongue manifests as erythema, fissuring, and atrophy of the papillae.23 Chronic lupus cheilitis is associated with cicatricial scarring and an increased risk of squamous carcinoma.22 Perianal mucosal involvement has occasionally been documented.23
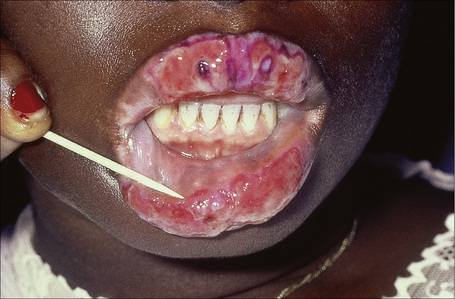
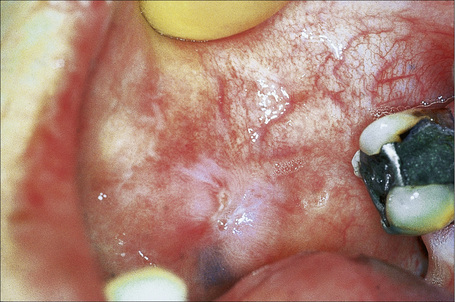
Fig. 17.16 Discoid lupus erythematosus: typical scarred lesion on the buccal mucosa.
By courtesy of R.A. Marsden, St George’s Hospital, London, UK.
Discoid lupus erythematosus has been described in association with osteopoikilosis, α1-antitrypsin deficiency, polyarteritis nodosa, in a patient with scleroderma and chronic hepatitis C virus infection, and as a reaction to tattoo.27–31
Verrucous (hypertrophic) discoid lupus erythematosus
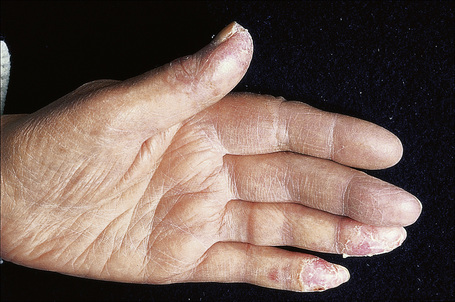
Verrucous (hypertrophic) DLE presents as warty, hyperkeratotic papules and plaques with a predilection for the face, scalp, mucous membranes of the lips, and upper limbs (Figs 17.18, 17.19).5,32–34 It affects approximately 2% of patients with chronic discoid disease.12 Although verrucous (hypertrophic) hyperkeratotic skin changes have been observed in patients with SLE, such cases are exceptional.34,35 Sometimes the palms and soles are affected and occasionally the nails (Figs 17.20, 17.21). Rare manifestations include nodular keratoacanthoma-like, squamous cell carcinoma-like, and hypertrophic lichen planus-like lesions on the arms and hands (Fig. 17.22).33,36 A variant with associated necrosis of the subcutaneous tissue is known as lupus erythematosus hypertrophicus et profundus.37
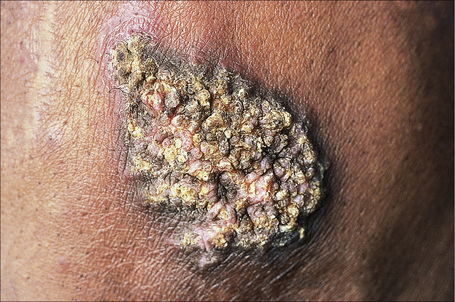
Fig. 17.18 Verrucous discoid lupus erythematosus: this lesion has a very warty appearance.
By courtesy of the Institute of Dermatology, London, UK.
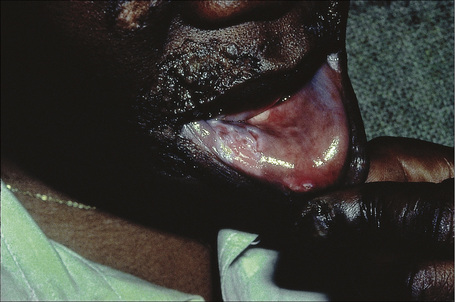
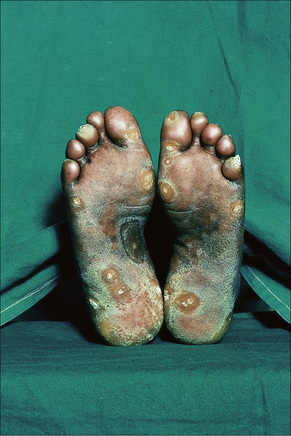
Fig. 17.20 Verrucous discoid lupus erythematosus: note the gross hyperkeratosis.
By courtesy of J. Newton Bishop, MD, St James’s University Hospital, Leeds, UK.
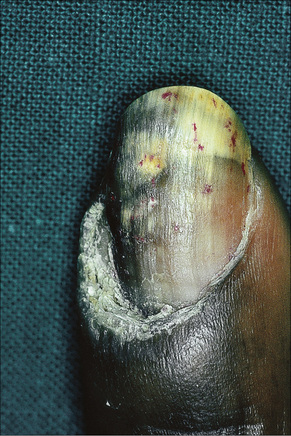
Fig. 17.21 Verrucous discoid lupus erythematosus: note the hypertrophic cuticle, pitting and onycholysis.
By courtesy of J. Newton Bishop, MD, St James’s University Hospital, Leeds, UK.
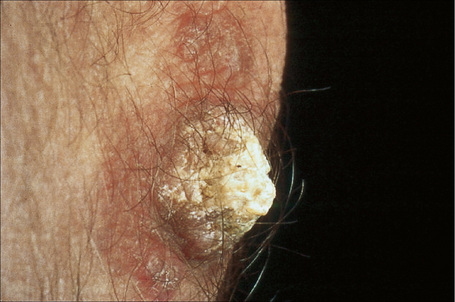
Fig. 17.22 Verrucous discoid lupus erythematosus: this example resembles a keratoacanthoma.
By courtesy of the Institute of Dermatology, London, UK.
Systemic symptoms are usually absent in patients with localized DLE.38 In the generalized form in which cutaneous plaques may be quite widespread, a small percentage may have Raynaud’s phenomenon and arthralgia (Figs 17.23–17.25). Laboratory abnormalities are more common in the generalized than in the localized variant.38 Anemia is not seen in localized DLE, but is sometimes a feature of the generalized variant. Leukopenia, a raised erythrocyte sedimentation rate (ESR), and hypergammaglobulinemia can occur in both.8 Antinuclear factor (diffuse pattern), a positive Wassermann reaction, and rheumatoid factor may also be features.8 Anti-double-stranded DNA (dsDNA) antibodies are detected in a minority of patients with disseminated DLE, and these patients frequently transform to the systemic disease; occasionally antibodies to single-stranded DNA (ssDNA) are present. Urinalysis and renal function tests are normal.
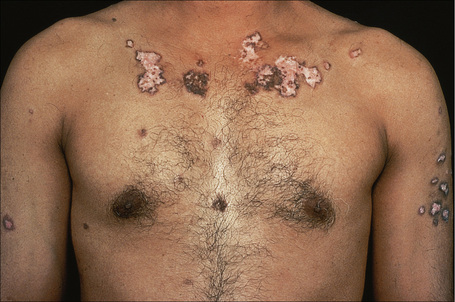
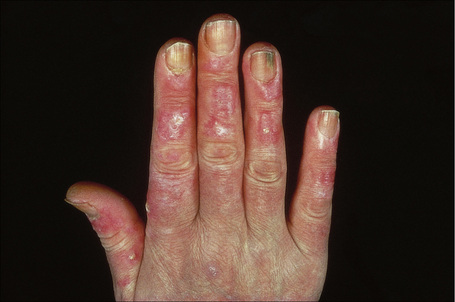
Fig. 17.24 Generalized discoid lupus erythematosus: note the extensive erythema and scaling.
By courtesy of R.A. Marsden, St George’s Hospital, London, UK.
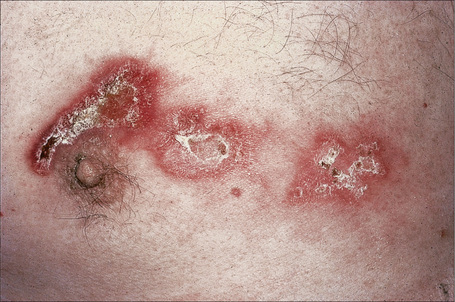
Fig. 17.25 Generalized discoid lupus erythematosus: in this patient, the chest is severely affected.
By courtesy of the Institute of Dermatology, London, UK.
Cutaneous squamous cell carcinoma and less often basal cell carcinoma may arise in patients with chronic lesions including the hypertrophic variant.19,21,33,39–43 Squamous cell carcinoma occurs predominantly in males, particularly affects the scalp, and is sometimes associated with early metastases.6,44
Lupus erythematosus tumidus
Lupus erythematosus tumidus is a distinctive subset of cutaneous lupus clinically presenting mainly in patients with DLE and rarely in patients with SLE. It is characterized by erythematous papules, plaques or even nodules with an urticarial appearance arising mainly on sun-exposed skin of the face, neck, and trunk. Scarring does not occur. This variant of lupus is discussed in more detail in chapter 8.
Chilblain lupus erythematosus
Chilblain lupus erythematosus (CHLE), which accounts for approximately 11% of DLE cases, develops during the winter months or following exposure to cold, wet, and damp conditions.45,46 Although CHLE appears most frequently in sporadic patients,47 familial occurrence has recently been reported.46,48,49 Patients with familial CHLE display mutations in the TREX1 gene located on chromosome 3p, encoding a DNA-specific 3′-5′ exonuclease 1.46,48,49 Autosomal dominant inheritance has been demonstrated in these patients.46,48,49 While familial CHLE generally presents in early childhood, sporadic CHLE usually affects middle-aged females.45,46,48,50 Patients develop itchy, painful, papuloerythematous or blue-purple plaques and nodules on the fingers, heels, and soles of the feet; the hands, calves, knees, knuckles, elbows, nose, and ears are less often affected (Fig. 17.26).6 Hyperkeratotic fissured lesions and ulcers are sometimes also present.50 CHLE may present with depigmentation mimicking vitiligo.51 Although patients usually develop chilblains many years after the typical discoid rash, lesions may develop simultaneously and sometimes the chilblains are the sole manifestation.50 Some patients with sporadic CHLE have an associated cryofibrinogenemia or cold agglutinin.6 About 20% of patients with sporadic CHLE develop SLE, particularly those who develop discoid and perniotic lesions simultaneously and those with DLE-erythema multiforme-like syndrome in addition to perniosis.6,45,47 None with the familial CHLE have progressed to SLE.47
Lupus erythematosus-erythema multiforme-like syndrome
The lupus erythematosus-erythema multiforme-like syndrome (Rowell’s syndrome) is rare.52–58 Patients, mostly middle-aged women, develop recurrent episodes of annular lesions on the limbs and, to a lesser extent, on the face, neck, chest, and mouth, in addition to the features of any variant of lupus erythematosus.6,52,59–61 Involvement of palms and soles is exceptional.62 Early lesions are erythematous papules, which become annular and may vesiculate at the edge. The condition usually heals without scarring, but in severe disease bullae may develop, become necrotic, and ulcerate. Patients with this variant also have erythrocyanosis, chilblains, and Raynaud’s phenomenon. Their serum contains speckled antinuclear factor, rheumatoid factor, and anti-Ro/La antibody.6,8,52,53 In rare cases, anti-Ro/La and rheumatoid factor are negative.63 Association with antiphospholipid syndrome has exceptionally been documented.64,65
Lupus erythematosus profundus
Lupus erythematosus profundus (panniculitis) is another uncommon variant that may develop in association with either DLE or SLE. This is discussed in detail in chapter 10.
DLE and chronic granulomatous disease
Occasionally, a DLE-like dermatosis develops in female carriers of X-linked chronic granulomatous disease.66–70 Mothers of affected children may occasionally show similar lesions, presenting with bluish-red, infiltrated, scaly papules on the face and hands, sometimes associated with photosensitivity.67 Additional features include recurrent aphthous-like ulcerative stomatitis and perniosis.
X-linked chronic granulomatous disease is inherited as a recessive trait, and patients (usually boys) have severe and recurrent infections due to defective neutrophil bactericidal activity. Heterozygous female carriers display a partial defect, indicated by diminished nitroblue tetrazolium reductions, but are usually free from infections. An autosomal recessive variant in which discoid lupus-like lesions may occur has also been documented.71 DLE-lesions have been reported in both carriers and chronic granulomatous disease patients; however, development of SLE in chronic granulomatous disease patients is extremely rare.72–76 Subacute cutaneous lupus erythematosus-like lesions and lupus tumidus may also occur.77,78 Discoid lupus erythematosus has also been described in non-X-linked hyper-IgM syndrome.79
Subacute cutaneous lupus erythematosus
Subacute cutaneous lupus erythematosus (SCLE) accounts for 5–10% of patients with lupus erythematosus.80–82 Approximately 50% have SLE as defined by the revised American Rheumatology Association diagnostic criteria (see below).83 SCLE predominates in Caucasians and is uncommon in Blacks, Koreans and Chinese.84 Females are affected more often than males (2.3:1) and the mean age at presentation is about 40 years.85 This variant of lupus occurs very rarely in children.86–88 Nail dystrophy affecting the majority of nails has been reported in a child with SCLE.88
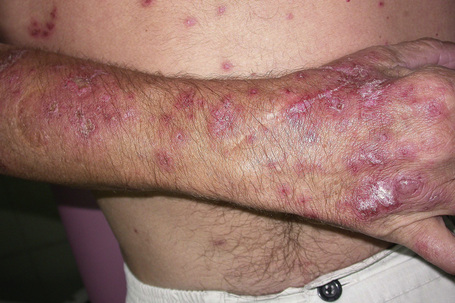
The eruption, which is often widely distributed, consists of symmetrical, nonscarring, and non-indurated erythematosquamous lesions (Fig. 17.27). Unique presentation along the lines of Blaschko has also been reported.89 Absence of induration has been regarded as a useful clinical discrimination feature between SCLE and DLE.84 In addition, the absence of follicular plugging, adherent scale, and dermal atrophy helps to distinguish the subacute lesion from the discoid variant.81 Pruritus is very rare.90 Lesions may become papulosquamous (psoriasiform) or annular, the latter coalescing to produce polycyclic and gyrate configurations (Fig. 17.28).81,91,92 In some patients both patterns are seen. Crusting and vesiculation are sometimes evident on the active border of the annular lesions.85 Pityriasiform and erythema multiforme-like lesions have also been described, as has occasional chronic leukoderma.81 Exceptionally, the disease may present as generalized poikiloderma93,94 or erythroderma.95 In addition, 15–30% of patients develop features of typical DLE, usually located on the scalp or face.1,81,83,96 The malar erythema of SLE is also sometimes evident (15%). Subtle hypopigmentation, telangiectasia, nonscarring alopecia, livedo reticularis, Raynaud’s phenomenon, and mucous membrane ulcers are additional features.81,85 Dystrophic calcification is exceptional.97 Cutaneous leukocytoclastic vasculitis is seen in a minority of patients and appears to be self-limited and not associated with a worsened prognosis.98
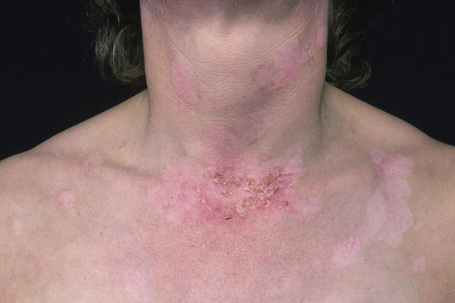
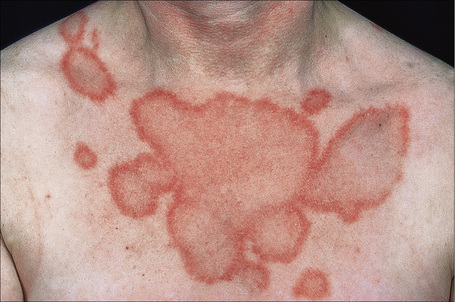
Photosensitivity is of major importance and lesions are therefore typically seen on the face, neck, upper part of back and chest, shoulders, extensor aspect of the arms, backs of hands, and fingers (Figs 17.29, 17.30).80,81,92
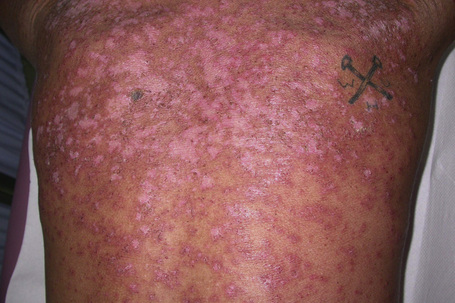
Fig. 17.29 Subacute cutaneous lupus erythematosus: in this patient, there is very extensive involvement.
By courtesy of Dr J.C. Pascual, Alicante, Spain.
Patients with SCLE have an increased incidence of human leukocyte antigen (HLA)-DR3 (75%), HLA-B8, and HLA-A1, and there is a significant association with inherited homozygous C2 and C4 deficiency.81,83,84,99–102 HLA-DR2 is also present at a higher frequency, particularly in those with papulosquamous rather than annular skin lesions.103 Antinuclear antibodies are found in approximately 50% of patients, while anti-Ro (SS-A) antibodies are present in approximately 65%, particularly those with annular polycyclic lesions.6,82,86,91,102 Anti-La (SS-B) antibodies are also often evident.5
The course of SCLE tends to be relatively benign, but systemic manifestations are quite common and may be severe.104 Severe extracutaneous disease appears to be more common in men with papulosquamous SCLE.104 The type of cutaneous lesion, however, has not always been proven to correlate with the severity of the extracutaneous manifestations.105 Renal disease has been reported to be uncommon but a recent study found its frequency to be as similar and equally severe as in SLE.85,106 Rarely, patients develop other more serious manifestations of SLE.83
Patients with SCLE have an increased incidence of both rheumatoid arthritis and Sjögren’s syndrome.82,107,108 Those with SCLE and Sjögren’s syndrome have high titers of anti-Ro antibodies, a high incidence of cutaneous vasculitis, and an increased risk of severe neuropsychiatric and pulmonary involvement.109 SCLE has been documented in association with porphyria cutanea tarda, hepatitis B virus infection, radiotherapy, squamous cell carcinoma of the head and neck region, breast, hepatocellular, and lung carcinoma.110–115 An exceptional association with inclusion body myositis and interstitial myositis with mitochondrial changes has also been documented.115,116 A relationship with lichen planus is exceptional.117 Clinically and histologically identical cases have been documented following therapy with hydrochlorothiazide, griseofulvin, antihistamines, terbinafine, calcium channel blockers, nifedipine, angiotensin-converting enzyme (ACE) inhibitors, proton pump inhibitors, interferon, phenytoin, bupropion, ticlopidine, capecitabine, lansoprazole, leflunomide, fluorouracil, leuprorelin, efalizumab, acebutolol, tamoxifen, docetaxel, and statin.118–147
Systemic lupus erythematosus
In addition to the variable cutaneous manifestations, lesions may be found in virtually any organ or system in the body in SLE.148–150 The guidelines of the American Rheumatology Association are valuable in establishing the diagnosis: any patient who has experienced four or more of the criteria, either serially or concurrently, is considered to have SLE (Table 17.2).151,152 The condition is characterized by a marked female predominance (9:1) and usually presents in the third, fourth, and fifth decades. There is a high incidence among Afro-Caribbeans, with a maximum prevalence of 1/150 females in Jamaica. In the United States the incidence is approximately 1/100.5
Table 17.2 Systemic lupus erythematosus: diagnostic guidelines of the American Rheumatology Association
| Criterion | Definition |
|---|---|
| Malar rash | Fixed erythema, flat or raised, over malar eminences tending to spare nasolabial folds |
| Discoid rash | Erythematous, raised patches with adherent keratotic scaling and follicular plugging; atrophic scarring may occur in old lesions |
| Photosensitivity | Skin rash as result of unusual reaction to sunlight (observed by physician or recounted by patient) |
| Oral ulcers | Oral or nasopharyngeal ulceration, usually painless, observed by physician |
| Arthritis | Nonerosive arthritis involving two or more peripheral joints, characterized by tenderness, swelling or effusion |
| Serositis | Pleurisy (convincing history of pleuritic pain or rub heard by physician or evidence of pleural effusion) Pericarditis (confirmed by ECG or rub or evidence of pericardial effusion) |
| Renal disorder | Persistent proteinuria (> 0.5 g/day or > 3 if quantification is not performed) Cellular casts (may be RBC, hemoglobin, granular, tubular or mixed) |
| Neurological disorder | Seizures (in absence of offending drugs or known metabolic derangements, e.g., uremia ketoacidosis or electrolyte imbalance) Psychosis (in absence of offending drugs or known metabolic derangements, e.g., uremia ketoacidosis or electrolyte imbalance) |
| Hematological disorder | Hemolytic anemia with reticulocytosis Leukopenia (< 4000/mm3 on two or more occasions) Lymphopenia (< 1500/mm3 on two or more occasions) Thrombocytopenia (< 1500/mm3 in absence of offending drug therapy) |
| Immunological disorder | Positive LE cell preparation Anti-DNA (antibody to native DNA in abnormal titer) Anti-SM (presence of antibody to SM nuclear antigen) False positive serologic test result for syphilis known to be positive for at least 6 months and confirmed by Treponema pallidum immobilization or fluorescent treponemal antibody absorption test |
| Antinuclear antibody | Abnormal ANA titer by immunofluorescence or equivalent assay at any time and in absence of drugs known to be associated with drug-induced lupus syndrome |
ANA, antinuclear antibody; LE, lupus erythematosus; RBC, red blood cell.
Reproduced with permission from Tan, E.M. et al. (1982) Arthritis and Rheumatism, 25, 1271–1277.
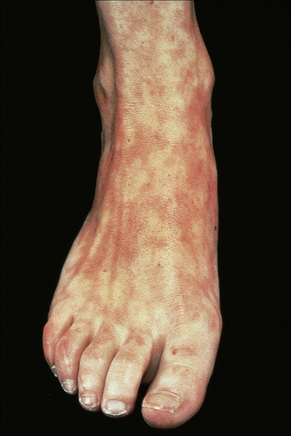
Cutaneous involvement occurs in 75–88% of patients.92 Lesions are highly polymorphic and may mimic many other dermatoses (Table 17.3). The ‘butterfly rash’ is typical and is a slightly scaly, sometimes edematous, erythema that is particularly distributed on the bridge of the nose and on the cheeks (Fig. 17.31). Photosensitivity is common, affecting more than 50% of patients, and erythematous or violaceous maculopapular eruptions may develop, particularly in white patients, at other light-exposed areas such as the ‘V’ of the neck, and the forearms.92 Sensitivity is towards both ultraviolet (UV) A and B light. Approximately 15% of patients, particularly Afro-Caribbeans, have lesions similar to those of DLE, apparently associated with a less severe disease and a lower frequency of renal involvement.3,10,92,148 SCLE-like features are present in 10–15% of patients.153 It has been suggested that patients with SLE and SCLE lesions have a more favorable prognosis.154
Table 17.3 Systemic lupus erythematosus: cutaneous manifestations
Reproduced from Moschella, S.L. (1989) Journal of Dermatology, 16, 417–428, with permission from Blackwell Publishing Ltd.
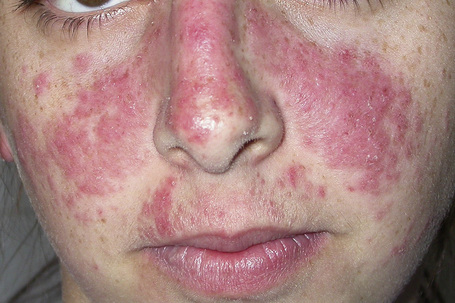
Fig. 17.31 Systemic lupus erythematosus: characteristic ‘butterfly erythema’ on the cheeks and nose.
By courtesy of Dr J.C. Pascual, Alicante, Spain.
Alopecia is important, occurring in about 20% of patients, and may be scarring or nonscarring. The nonscarring lesions, which are more common, often constitute a fairly diffuse hair loss occurring as a non-specific response to stress (telogen effluvium).92 Fractured frontal hairs are characteristic.19
Raynaud’s phenomenon occurs in 10–40% of patients.92 Purpura and ecchymoses are common and may be partly due to corticosteroid therapy, but thrombocytopenia and immune complex-mediated vasculitis also play a role in the pathogenesis. Vasculitis, which occurs in up to 30% of patients, may also result in infarcts, ulcers, digital nodules, scars, and gangrene (Figs 17.32–17.34).92 Livedo reticularis that particularly affects the arms and thighs and periarticular sites may be a presenting feature in up to 10% of patients, and the changes of atrophie blanche have been documented occasionally (livedoid vasculitis) (Fig. 17.35).92 Livedo reticularis is often a feature of the anticardiolipin syndrome and presenting lesions identical to those seen in Degos’ disease (malignant atrophic papulosis) have been described occasionally.155,156 Vasculitic features correlate with renal and central nervous system involvement.92 Urticaria is frequently found.157
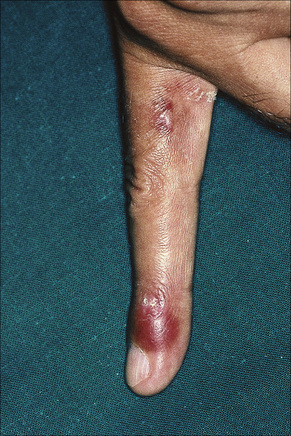
Fig. 17.32 Systemic lupus erythematosus: erythematous vasculitic lesion on the fingertip.
By courtesy of M.M. Black, MD, St Thomas’ Hospital, London, UK.
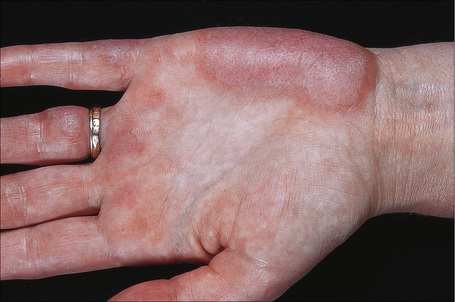
Fig. 17.33 Systemic lupus erythematosus: involvement of the palms is rare and may be vasculitic.
By courtesy of the Institute of Dermatology, London, UK.
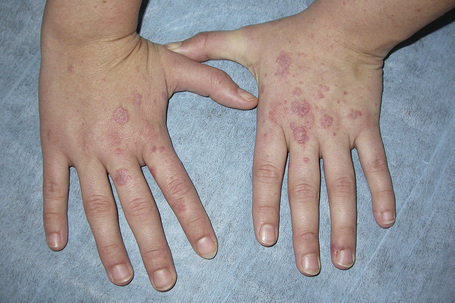
Erythromelalgia, a burning sensation accompanied by erythema following exposure to heat, has also been noted.21 Localized and diffuse hyperpigmentation and urticarial lesions, including urticarial vasculitis, are less commonly found. Digital manifestations, in addition to infarction and ulceration, include periungual and knuckle erythema, nail fold telangiectases, cuticular lesions, and splinter hemorrhages. Telangiectases may also be seen on the fingertips and palms.91 Some patients develop red lunulae.158,159 The associated erythema multiforme-like and perniotic lesions are described above. Involvement of the mucous membranes occurs in about 10% of patients: painless ulceration is most common, but other features include erythema, petechiae, erosions, and hemorrhage. The central part of the hard palate, lips, and buccal mucosa are particularly affected.24,156,160
Additional lesions that are occasionally seen include rheumatoid-like nodules, dermal mucinosis, bullous variants, thrombotic thrombocytopenic purpura, mixed cryoglobulinemia, and soft tissue calcifications.5,19,161,162 Vesicles and bullae in SLE may complicate extreme basal cell hydropic degeneration, represent coexpression of an autoimmune bullous dermatosis, or signify a specific dermatitis herpetiformis-like eruption (bullous dermatosis of SLE). There is a recognized association with porphyria cutanea tarda.163
Approximately 5–10% of patients with SLE are antinuclear antibody negative. This seems to correlate with a specific subtype. Patients are usually HLA-DR3 positive and have Sjögren’s syndrome and an increased incidence of pulmonary involvement, psychiatric manifestations, cutaneous vasculitis, and hypergammaglobulinemic purpura.5
Non-specific symptoms are common and include chronic tiredness, weight loss, fever, malaise, and weakness. Arthralgia and myalgia occur in 90% of patients. The myalgia may be disabling, but objective muscle weakness is rare.164 Similarly, although arthralgia may be marked, there is usually little clinical evidence of joint damage. Effusions are common.148 Approximately 25% of patients have frank arthritis, either a migratory polyarthritis or a chronic progressive polyarthritis with deformity. Patients sometimes develop avascular bone necrosis due either to the primary disease or to steroid therapy.148,164 Very rarely, involvement of the tendons is associated with the development of contractures.
Lesions of the cardiovascular system manifest as cardiomegaly, pericarditis, pericardial effusion and/or endocarditis (Libman-Sacks valvulitis).165 In SLE, the mitral valve is predominantly affected, but patients with the lupus anticoagulant syndrome appear to have a particular risk of developing aortic valve disease.165,166 Conduction defects and congestive cardiac failure are additional features.
Respiratory involvement may present as pleurisy, with or without effusion, bacterial pneumonia or, very rarely, lupus pneumonitis.164 Pulmonary hypertension has been estimated to be present in 0.5–14% of SLE patients, especially those associated with antiphospholipid antibodies.167
Involvement of the central nervous system is an important cause of morbidity and mortality.148,164 It may affect up to 40% of patients.6 Encephalitis, meningitis, vasculitis, and coagulation defects give rise to a wide range of clinical manifestations, including convulsions, hemiplegias, chorea, and psychoses.19 Convulsions and coma are an indication of severe involvement and portend a grave outcome. Peripheral neuritis affects up to 12% of patients; ocular lesions, including conjunctivitis, fundal hemorrhages, and cotton wool exudates, occur in 25%.19
Renal manifestations develop in approximately 45% of patients, and progressive renal involvement is an important cause of morbidity and mortality (nephrotic syndrome and lupus glomerulonephritis). Evidence of active renal disease includes the presence in the urine of more than five red blood cells/high-power field.164 Proteinuria of greater than 1 g/24 hours, oval fat bodies, granular, hyaline, and red blood cell casts may also be detected.19
Gastrointestinal manifestations are uncommon; the most important is esophageal involvement leading to loss of peristalsis and dilatation reminiscent of that seen in scleroderma.164
Laboratory investigation commonly reveals anemia, leukopenia, lymphopenia, thrombocytopenia, and a raised ESR. False-positive reactions to reagin and treponemal tests for syphilis are common, 10–20% of patients are positive for the Coombs’ test and rheumatoid factor, and 10–50% have circulating anticoagulants.148
While lupus erythematosus (LE) cell preparation used to be the basic screening test for SLE, it has now been superseded by testing for antinuclear factor. This and other autoantibodies will be discussed further under pathogenesis. Serum complement levels are often low in patients with active disease (CH50 and C3); estimations of C3 levels are of particular value in following disease activity.5,19
An association between SLE and inherited complement deficiency, involving the early components of the classic pathway including C1r, C1s, C1q, C4, and C2, has been described.168–170 Deficiency of C2 is inherited in an autosomal recessive fashion; up to 60% of homozygotes develop lupus erythematosus characterized by an erythematous, papulosquamous, SCLE-like photosensitive dermatosis, a low incidence of renal involvement, and arthralgia. Additional features may include urticarial vasculitis, malar erythema and nail fold abnormalities.100 Discoid lesions are sometimes evident, and recently a patient with C2 and C4 deficiency and LE panniculitis has been documented.12,171 Patients are, however, often (but not invariably) negative for antinuclear factor and manifest a negative lupus band test on unaffected skin. There is an association with HLA-DR2, and over 60% of patients possess anti-Ro antibodies.80
Numerous drugs have been implicated in the induction of SLE including:
Transient skin and serological manifestations of SLE have been documented in two children with trichophyton mentagrophytes infection.211 The disease has also been linked to hepatitis B vaccination and with exposure to insecticides.232–235
Sjögren’s syndrome coexists in approximately 10–20% of patients with SLE.91 SLE has also been described in association with scleroderma, morphea, rheumatoid arthritis, eosinophilic fasciitis, dermatomyositis, lichen sclerosus, pemphigus (including pemphigus vulgaris, foliaceous, and paraneoplastic pemphigus), hypergammaglobulinemia of Waldenström, dermatitis herpetiformis, ulcerative colitis, alopecia areata, autoimmune thyroiditis, myasthenia gravis, acanthosis nigricans, Sweet’s syndrome, porphyria, gout, sarcoidosis, psoriasis, lichen planus, and cutaneous T-cell lymphoma. It is likely that many of these associations are chance associations except for those diseases with an autoimmune basis.163, 236–260
Neonatal lupus erythematosus
Neonatal lupus erythematosus is very rare, occurring in approximately 1 in 20 000 live births261 and most commonly presents in female infants.261–263 It is associated with maternal anti-Ro (SS-A) antibodies (95%) and/or anti-La (SS-B) antibodies.262,263 Anti-U1-ribonucleoprotein (RNP) antibodies may also be present.264–268 Anticardiolipin antibodies have been described in a single case.269 Transplacental transfer of anti-Ro and anti-La antibodies results in a SCLE-like eruption consisting of erythematous annular scaly plaques that particularly affect the periorbital region (‘owl-eye’ or ‘eye-mask’) and scalp, and to a lesser extent, the trunk and extremities (Figs 17.36, 17.37).5 These cutaneous lesions often appear after exposure to ultraviolet light. Photosensitivity in neonatal lupus erythematosus patients appears to show racial differences, being more frequent in Caucasian and Americans than in Japanese infants.270 Crusted lesions and cutis marmorata telangiectasia congenita are additional features in some cases.271,272 Exceptionally, discoid lesions, panniculitis (lupus profundus), multiple morphea, erosions, and alopecia may also occur.273–275 Atrophy and scarring are very rare but residual hypopigmentation and telangiectasia are present in about 25% of patients.276 Babies in subsequent pregnancies may present manifestations of the disease277,278 and the overall recurrence rate for neonatal lupus-associated cardiac disease is 17%.279
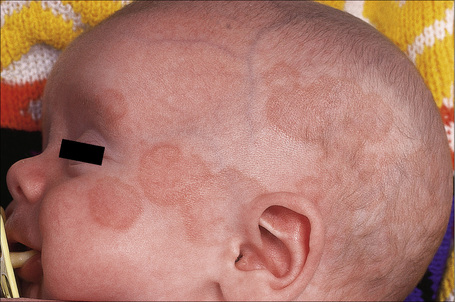
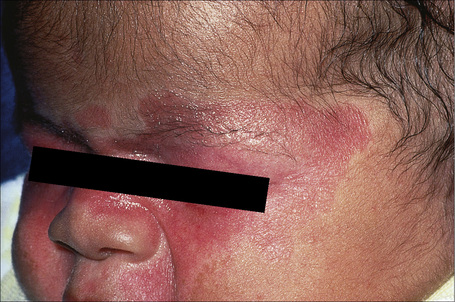
Neonatal lupus erythematosus is often associated with complete heart block, and sometimes liver disease (cholestatic hepatitis), splenomegaly, thrombocytopenia, leukopenia, and anemia are also evident.262,275 Circulating autoantibodies to annexin A6 were detected in a single patient with neonatal lupus erythematosus-related dilated cardiomyopathy.280 Multiple mucocutaneous and visceral hemangiomas have been reported in a single patient.281 The mortality associated with cardiac involvement is around 19%.277 Severe hematological and liver involvement without evidence of cutaneous or cardiac involvement has exceptionally been reported.282 By 6 months of age the cutaneous, but not the cardiac manifestations, usually fade. The levels of maternal autoantibodies in the infant decrease quickly during the first weeks of life and generally become undetectable by the age of 1 year.283
The disease also occurs in identical and nonidentical twins.284 A single case has been described in association with Turner’s syndrome.285 Neonates who survive until infancy have a fairly good prognosis, but there is an overall mortality of about 10% due to heart disease.262 Mothers of affected infants are commonly asymptomatic initially but they may have systemic lupus, Sjögren’s syndrome, leukocytoclastic vasculitis or an overlap syndrome.286
A number of mothers who present with no symptoms, often subsequently develop evidence of connective tissue disease, particularly systemic lupus (in about 20% of cases) and Sjögren’s syndrome.262
Pathogenesis and histological features
Despite enormous research efforts, the precise etiology and pathogenesis are unknown, but the condition is certainly multifactorial.287,288 Lupus erythematosus is characterized by B-cell hyperactivity in association with defective suppressor T-cell function. Patients develop a wide range of autoantibodies, many of which result in immune complexes with resultant systemic manifestations including vasculitis and glomerulonephritis. In addition to immunological factors, familial, genetic, and hormonal influences play a part. It is believed that in lupus erythematosus there is a genetic predisposition; sex-associated and environmental factors are necessary to promote the development of the disease.
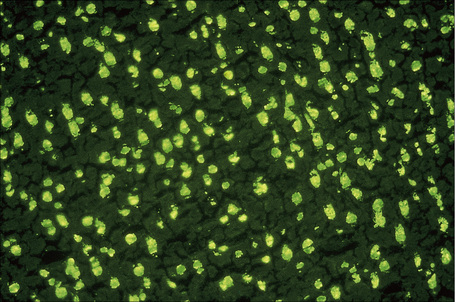
Fig. 17.38 Systemic lupus erythematosus: homogeneous antinuclear antibody in rat liver.
By courtesy of G. Swana, MD, St Thomas’ Hospital, London, UK.
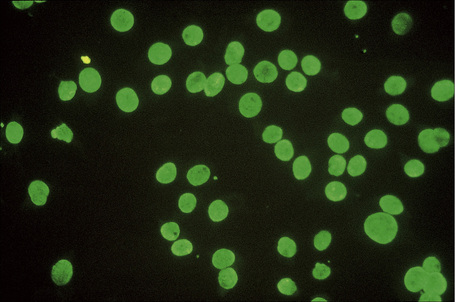
Fig. 17.39 Systemic lupus erythematosus: high-power view.
By courtesy of G. Swana, MD, St Thomas’ Hospital, London, UK.
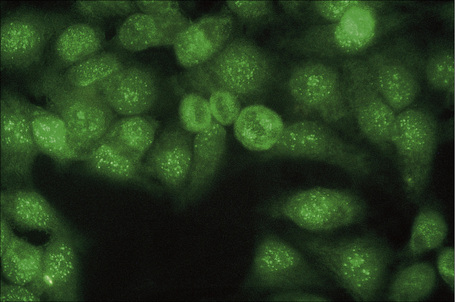
Fig. 17.40 Systemic lupus erythematosus: speckled antinuclear antibody – HEP II.
By courtesy of G. Swana, MD, St Thomas’ Hospital, London, UK.
Patients with lupus erythematosus develop antibodies to a variety of nuclear and cytoplasmic antigens; these autoantibodies, both singly and in combination, have varying associations, which may allow a prediction of (to some extent, at least) the course of the disease in individual patients (Table 17.4). Antibodies to native (double-stranded) DNA (nDNA) are pathognomonic for idiopathic (classic) SLE (Fig. 17.41).6,19 They are rarely seen in the drug-induced variant and are only very occasionally present in patients with DLE.2 The presence of anti-nDNA antibody in association with hypocomplementemia is indicative of active disease and is often accompanied by severe renal involvement.
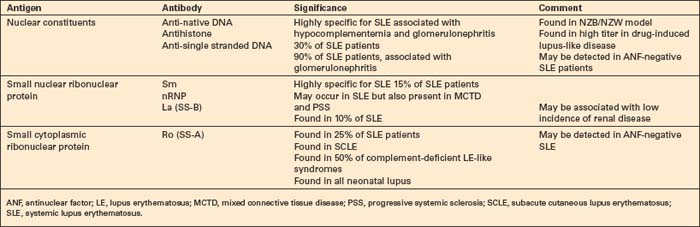
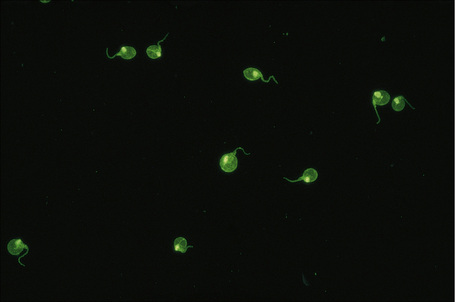
Fig. 17.41 Systemic lupus erythematosus: anti-dsDNA (Crithidia luciliae).
By courtesy of G. Swana, MD, St Thomas’ Hospital, London, UK.
Antihistone antibodies may be found in approximately 30% of patients with idiopathic SLE, but are particularly associated with the drug-induced variant.289 The latter is caused by a wide variety of drugs, including hydralazine, procainamide hydrochloride, phenytoin, and isoniazid.14,290,291 Symptoms develop in up to 20% of patients taking procainamide and antinuclear antibodies are present in 50%.291 Procainamide-induced SLE-like syndrome is characterized by the presence of leukocyte-specific antinuclear antibody.291 Clinical features include malaise, pneumonitis with pleural effusion, arthralgia, arthritis, and serositis; renal, central nervous system, and cutaneous lesions are less common and are usually mild. Although antinuclear antibodies are often present, anti-DNA antibodies are not formed in most cases of drug-induced lupus.164 Drug-induced lupus usually, but not invariably, undergoes remission on withdrawal of the drug.291
Antibodies to ssDNA occur in 90% of patients with classic SLE and in approximately 20% of patients with disseminated DLE, and may also be found in other connective tissue diseases.10 Their presence in disseminated DLE correlates with an increased risk of developing SLE.10 Patients with SLE who are negative for antinuclear antibody may have anti-ssDNA antibodies in their serum.6 Antibodies to the soluble nuclear antigen Sm are highly specific for SLE.289 They may also be found in a group of patients with good prognosis who are positive for antinuclear factor, but anti-nDNA-negative, and who have mild nonprogressive glomerulonephritis, hypocomplementemia, mild central nervous system involvement, and conspicuous cutaneous eruptions.
Antibodies to ribonucleoprotein are detected in 23% of patients with SLE, but are much more commonly associated with mixed connective tissue disease: they may also be found in patients with progressive systemic sclerosis.289 Anti-La (SS-B) antibodies are detected in the serum of 10% of patients with SLE. Anti-La antibodies, which are often present in Sjögren’s syndrome, are almost invariably accompanied by anti-Ro (SS-A) antibodies.289 The latter are particularly associated with SCLE, complement-deficient lupus erythematosus, circulating IgG or IgM anticoagulant, and neonatal lupus erythematosus. About 15–20% of patients with SLE have detectable antineutrophil cytoplasmic antibodies (ANCA).292
The circulating anticoagulant (antiphospholipid or lupus anticoagulant) is associated with paradoxical thrombosis, spontaneous abortion, premature labor, intrauterine death, labile hypertension, cutaneous necrosis, gangrene, ecchymoses, purpura, leg ulcers, atrophie blanche, livedo reticularis, and false-positive syphilis serology – the lupus anticoagulant syndrome (Table 17.5).289,293–304 The lupus anticoagulant syndrome is more accurately known as the antiphospholipid syndrome because not all patients have associated SLE. It is due to the presence of circulating antiphospholipid antibodies which inhibit coagulation in vitro, but more importantly are associated with a greatly increased risk of thrombotic phenomena affecting both arteries and veins (Figs. 17.42, 17.43). About 50% of patients with SLE present with antiphospholipid antibodies but only half of these will develop manifestations of the disease.305 Presentation in the skin may also be with papules and nodules or lesions resembling pyoderma gangrenosum.306,307 Interestingly, patients with antiphospholipid antibodies and SLE appear to be more at risk of developing anetoderma.308–310 The incidence, however, is not high. Rarely, patients develop reactive angioendotheliomatosis.311,312 Systemic involvement includes deep venous thrombosis often complicated by pulmonary embolism, arterial occlusion with resultant strokes, transient ischemic attacks, multi-infarct dementia, myocardial infarction, and gangrene.289
Table 17.5 Diagnostic criteria for the antiphospholipid syndrome
| Group | Criteria |
|---|---|
| I. Clinical conditions | |
| II. Laboratory* | |
| III. Other clinical conditions | < div class='tao-gold-member'> Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|