Hypnotics and Sedatives
The CNS depressants discussed in this chapter include benzodiazepines, other benzodiazepine receptor agonists (the “Z compounds”), barbiturates, and sedative-hypnotic agents of diverse chemical structure. Older sedative-hypnotic drugs depress the CNS in a dose-dependent fashion, progressively producing a spectrum of responses from mild sedation to coma and death. A sedative drug decreases activity, moderates excitement, and calms the recipient, whereas a hypnotic drug produces drowsiness and facilitates the onset and maintenance of a state of sleep that resembles natural sleep in its electroencephalographic characteristics and from which the recipient can be aroused easily.
Sedation is a side effect of many drugs that are not general CNS depressants (e.g., antihistamines and antipsychotic agents). Although such agents can intensify the effects of CNS depressants, they usually produce more specific therapeutic effects at concentrations far lower than those causing substantial CNS depression. The benzodiazepine sedative-hypnotics resemble such agents; although coma may occur at very high doses, neither surgical anesthesia nor fatal intoxication is produced by benzodiazepines in the absence of other drugs with CNS-depressant actions; an important exception is midazolam, which has been associated with decreased tidal volume and respiratory rate. Moreover, specific antagonists of benzodiazepines exist. This constellation of properties sets the benzodiazepine receptor agonists apart from other sedative-hypnotic drugs and imparts a measure of safety such that benzodiazepines and the newer Z compounds have largely displaced older agents for the treatment of insomnia and anxiety.
The sedative-hypnotic drugs that do not specifically target the benzodiazepine receptor belong to a group of agents that depress the CNS in a dose-dependent fashion, progressively producing calming or drowsiness (sedation), sleep (pharmacological hypnosis), unconsciousness, coma, surgical anesthesia, and fatal depression of respiration and cardiovascular regulation. They share these properties with a large number of chemicals, including general anesthetics (see Chapter 19) and alcohols, most notably ethanol (see Chapter 23).
BENZODIAZEPINES
All benzodiazepines in clinical use promote the binding of the major inhibitory neurotransmitter γ-aminobutyric acid (GABA) to the GABAA receptor, a multi-subunit, ligand-gated chloride channel. GABA binding induces the Cl– current through these channels (see Figure 14–6).
Pharmacological data suggest heterogeneity among sites of binding and action of benzodiazepines; different subunit combinations comprise the GABA-gated chloride channels expressed in different neurons. Receptor subunit composition appears to govern the interaction of various allosteric modulators with these channels, giving hope to efforts to find agents displaying different combinations of benzodiazepine-like properties that reflect selective actions on 1 or more subtypes of GABAA receptors. A number of distinct mechanisms of action are thought to contribute to the sedative-hypnotic, muscle-relaxant, anxiolytic, and anticonvulsant effects of the benzodiazepines, and specific subunits of the GABAA receptor are responsible for specific pharmacological properties of benzodiazepines. While only the benzodiazepines used primarily for hypnosis are discussed in detail, this chapter describes the general properties of the group and important differences among individual agents (see also Chapters 15 and 21).
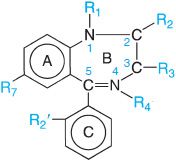
Benzodiazepine refers to the portion of the this structure composed of a benzene ring (A) fused to a 7-membered diazepine ring (B). Because all the important benzodiazepines contain a 5-aryl substituent (ring C) and a 1,4-diazepine ring, the term has come to mean the 5-aryl-1,4-benzodiazepines. Numerous modifications in the structure of the ring systems and substituents have yielded compounds with similar activities, including flumazenil (ROMAZICON, in which ring C is replaced with a keto function at position 5 and a methyl substituent is added at position 4), a benzodiazepine receptor antagonist. A large number of nonbenzodiazepine compounds compete with classic benzodiazepines and flumazenil for binding at specific sites in the CNS (e.g., β-carbolines, zolpidem, eszopiclone).
PHARMACOLOGICAL PROPERTIES
Virtually all effects of the benzodiazepines result from their actions on the CNS. The most prominent of these effects are sedation, hypnosis, decreased anxiety, muscle relaxation, anterograde amnesia, and anticonvulsant activity. Only 2 effects of these drugs result from peripheral actions: coronary vasodilation, seen after intravenous administration of therapeutic doses of certain benzodiazepines, and neuromuscular blockade, seen only with very high doses.
CNS. While benzodiazepines depress activity at all levels of the neuraxis, some structures are affected preferentially. The benzodiazepines do not produce the same degrees of neuronal depression produced by barbiturates and volatile anesthetics. All the benzodiazepines have similar pharmacological profiles. Nevertheless, the drugs differ in selectivity, and the clinical usefulness of individual benzodiazepines thus varies considerably.
As the dose of a benzodiazepine is increased, sedation progresses to hypnosis and then to stupor. The clinical literature often refers to the “anesthetic” effects and uses of certain benzodiazepines, but the drugs do not cause a true general anesthesia because awareness usually persists, and immobility sufficient to allow surgery cannot be achieved. Nonetheless, at “preanesthetic” doses, there is amnesia for events subsequent to administration of the drug. Despite to separate the anxiolytic actions of benzodiazepines from their sedative-hypnotic effects, distinguishing between these behaviors is problematic. Measurements of anxiety and sedation are difficult in humans, and the validity of animal models for anxiety and sedation is uncertain. The existence of multiple benzodiazepine receptors may explain in part the diversity of pharmacological responses in different species.
Tolerance to Benzodiazepines. Studies on tolerance in laboratory animals often are cited to support the belief that disinhibitory effects of benzodiazepines are distinct from their sedative-ataxic effects. Although most patients who ingest benzodiazepines chronically report that drowsiness wanes over a few days, tolerance to the impairment of some measures of psychomotor performance (e.g., visual tracking) usually is not observed. Whether tolerance develops to the anxiolytic effects of benzodiazepines remains a subject of debate. Many patients can maintain themselves on a fairly constant dose; increases or decreases in dosage appear to correspond with changes in problems or stresses. Conversely, some patients either do not reduce their dosage when stress is relieved or steadily escalate dosage. Such behavior may be associated with the development of drug dependence (see Chapter 24).
Some benzodiazepines induce muscle hypotonia without interfering with normal locomotion and can decrease rigidity in patients with cerebral palsy. Clonazepam in nonsedative doses does cause muscle relaxation, but diazepam and most other benzodiazepines do not. Tolerance occurs to the muscle relaxant and ataxic effects of these drugs.
Clonazepam, nitrazepam, and nordazepam have more selective anticonvulsant activity than most other benzodiazepines. Benzodiazepines also suppress photic seizures in baboons and ethanol-withdrawal seizures in humans. However, the development of tolerance to the anticonvulsant effects has limited the usefulness of benzodiazepines in the treatment of recurrent seizure disorders in humans (see Chapter 21).
Although analgesic effects of benzodiazepines have been observed in experimental animals, only transient analgesia is apparent in humans after intravenous administration. Such effects actually may involve the production of amnesia. Unlike barbiturates, benzodiazepines do not cause hyperalgesia.
Effects on the Electroencephalogram (EEG) and Sleep Stages. The effects of benzodiazepines on the waking EEG resemble those of other sedative-hypnotic drugs. Alpha activity is decreased, but there is an increase in low-voltage fast activity. Tolerance occurs to these effects. With respect to sleep, some differences in the patterns of effects exerted by the various benzodiazepines have been noted, but benzodiazepine use usually imparts a sense of deep or refreshing sleep. Benzodiazepines decrease sleep latency, especially when first used, and diminish the number of awakenings and the time spent in stage 0 (a stage of wakefulness). Time in stage 1 (descending drowsiness) usually is decreased, and there is a prominent decrease in the time spent in slow-wave sleep (stages 3 and 4). Most benzodiazepines increase the time from onset of spindle sleep to the first burst of rapid-eye-movement (REM) sleep; the time spent in REM sleep usually is shortened, however, the number of cycles of REM sleep usually is increased, mostly late in the sleep time. Zolpidem and zaleplon suppress REM sleep to a lesser extent than do benzodiazepines and thus may be superior to benzodiazepines for use as hypnotics.
Despite the shortening of stage 4 and REM sleep, benzodiazepine administration typically increases total sleep time, largely by increasing the time spent in stage 2 (which is the major fraction of non-REM sleep). The effect is greatest in subjects with the shortest baseline total sleep time. In addition, despite the increased number of REM cycles, the number of shifts to lighter sleep stages (1 and 0) and the amount of body movement are diminished. Nocturnal peaks in the secretion of growth hormone, prolactin, and luteinizing hormone are not affected. During chronic nocturnal use of benzodiazepines, the effects on the various stages of sleep usually decline within a few nights. When such use is discontinued, the pattern of drug-induced changes in sleep parameters may “rebound,” and an increase in the amount and density of REM sleep may be especially prominent. If the dosage has not been excessive, patients usually will note only a shortening of sleep time rather than an exacerbation of insomnia.
MOLECULAR TARGETS FOR BENZODIAZEPINE ACTIONS IN THE CNS. Benzodiazepines act at GABAA receptors by binding directly to a specific site that is distinct from that of GABA binding (see Figure 14–6). Unlike barbiturates, benzodiazepines do not activate GABAA receptors directly; rather, benzodiazepines act allosterically by modulating the effects of GABA. Benzodiazepines and GABA analogs bind to their respective sites on brain membranes with nanomolar affinity. Benzodiazepines modulate GABA binding, and GABA alters benzodiazepine binding in an allosteric fashion.
Benzodiazepines and related compounds can act as agonists, antagonists, or inverse agonists at the benzodiazepine-binding site on GABAA receptors. Agonists at the binding site increase, and inverse agonists decrease, the amount of chloride current generated by GABAA-receptor activation. Agonists at the benzodiazepine binding site shift the GABA concentration-response curve to the left, whereas inverse agonists shift the curve to the right. Both these effects are blocked by antagonists at the benzodiazepine binding site. In the absence of an agonist or inverse agonist for the benzodiazepine binding site, an antagonist for this binding site does not affect GABAA receptor function. One such antagonist, flumazenil, is used clinically to reverse the effects of high doses of benzodiazepines. The behavioral and electrophysiological effects of benzodiazepines also can be reduced or prevented by prior treatment with antagonists at the GABA binding site (e.g., bicuculline).
Each GABAA receptor is believed to consist of a pentamer of homologous subunits. Thus far 16 different subunits have been identified and classified into 7 subunit families. The exact subunit structures of native GABA receptors still are unknown; most GABA receptors are likely of α, β, and γ subunits that co-assemble with some uncertain stoichiometry. The multiplicity of subunits generates heterogeneity in GABAA receptors and is responsible, at least in part, for the pharmacological diversity in benzodiazepine effects in behavioral, biochemical, and functional studies. An understanding of which GABAA receptor subunits are responsible for particular effects of benzodiazepines in vivo is emerging from KO animal studies. The attribution of specific behavioral effects of benzodiazepines to individual receptor subunits will, hopefully, aid in the development of new compounds exhibiting fewer undesired side effects.
GABAA Receptor-Mediated Electrical Events. Benzodiazepines exert their major actions by increasing the gain of inhibitory neurotransmission mediated by GABAA receptors. Enhancement of GABA-induced chloride currents by benzodiazepines results primarily from an increase in the frequency of bursts of Cl– channel opening produced by submaximal amounts of GABA. At therapeutically relevant concentrations, benzodiazepines potentiate inhibitory synaptic transmission measured after stimulation of afferent fibers.
The remarkable safety of the benzodiazepines likely relates to the fact that their effects in vivo depend on the presynaptic release of GABA; in the absence of GABA, benzodiazepines have no effects on GABAA receptor function. This is in distinction to barbiturates, which also enhance the effects of GABA at low concentrations, but, in addition, directly activate GABA receptors at higher concentrations, which can lead to profound CNS depression.
The behavioral and sedative effects of benzodiazepines can be ascribed in part to potentiation of GABA-ergic pathways that serve to regulate the firing of monoamine-containing neurons known to promote behavioral arousal and to be important mediators of the inhibitory effects of fear and punishment on behavior. Finally, inhibitory effects on muscular hypertonia or the spread of seizure activity can be rationalized by potentiation of inhibitory GABA-ergic circuits at various levels of the neuraxis. The magnitude of the effects produced by benzodiazepines varies widely depending on such factors as the types of inhibitory circuits that are operating, the sources and intensity of excitatory input, and the manner in which experimental manipulations are performed and assessed. Accordingly, benzodiazepines markedly prolong the period after brief activation of recurrent GABA-ergic pathways during which neither spontaneous nor applied excitatory stimuli can evoke neuronal discharge; this effect is reversed by the GABAA receptor antagonist bicuculline (see Figure 14–5).
Respiration. Hypnotic doses of benzodiazepines are without effect on respiration in normal subjects, but special care must be taken in the treatment of children and individuals with impaired hepatic function. At higher doses, such as those used for preanesthetic medication or for endoscopy, benzodiazepines slightly depress alveolar ventilation and cause respiratory acidosis as the result of a decrease in hypoxic rather than hypercapnic drive; these effects are exaggerated in patients with chronic obstructive pulmonary disease (COPD), and alveolar hypoxia and CO2 narcosis may result. These drugs can cause apnea during anesthesia or when given with opioids. Patients severely intoxicated with benzodiazepines only require respiratory assistance when they also have ingested another CNS-depressant drug, most commonly ethanol.
In contrast, hypnotic doses of benzodiazepines may worsen sleep-related breathing disorders by adversely affecting control of the upper airway muscles or by decreasing the ventilatory response to CO2. The latter effect may cause hypoventilation and hypoxemia in some patients with severe COPD. In patients with obstructive sleep apnea (OSA), hypnotic doses of benzodiazepines may decrease muscle tone in the upper airway and exaggerate the impact of apneic episodes on alveolar hypoxia, pulmonary hypertension, and cardiac ventricular load. Benzodiazepines may promote the appearance of episodes of apnea during REM sleep (associated with decreases in oxygen saturation) in patients recovering from a myocardial infarction; however, no impact of these drugs on survival of patients with cardiac disease has been reported.
Cardiovascular System. The cardiovascular effects of benzodiazepines are minor in normal subjects except in severe intoxication. In preanesthetic doses, all benzodiazepines decrease blood pressure and increase heart rate. With midazolam, effects appear to be secondary to a decrease in peripheral resistance; with diazepam, effects are secondary to a decrease in left ventricular work and cardiac output. Diazepam increases coronary flow, possibly by an action to increase interstitial concentrations of adenosine, and the accumulation of this cardiodepressant metabolite also may explain the negative inotropic effects of the drug. In large doses, midazolam decreases cerebral blood flow and oxygen assimilation considerably.
GI Tract. Benzodiazepines are thought by some gastroenterologists to improve a variety of “anxiety-related” GI disorders. Diazepam markedly decreases nocturnal gastric secretion in humans. Other drug classes are considerably more effective in acid-peptic disorders (see Chapter 45).
ABSORPTION, FATE, AND EXCRETION. All the benzodiazepines are absorbed completely (clorazepate is decarboxylated rapidly in gastric juice to N-desmethyldiazepam [nordazepam], which subsequently is absorbed completely). Drugs active at the benzodiazepine receptor may be divided into 4 categories based on their elimination t1/2:
• Ultra-short-acting benzodiazepines
• Short-acting agents (t1/2 6 h), including triazolam, the non-benzodiazepine zolpidem (t1/2 2 h), and eszopiclone (t1/2 5-6 h)
• Intermediate-acting agents (t1/2 6-24 h), including estazolam and temazepam
• Long-acting agents (t1/2 > 24 h), including flurazepam, diazepam, and quazepam
Flurazepam itself has a short t1/2 (~2.3 h), but a major active metabolite, N-des-alkyl-flurazepam, is long-lived (t1/2 47-100 h); such features complicate the classification of individual benzodiazepines.
The benzodiazepines and their active metabolites bind to plasma proteins. The extent of binding correlates strongly with lipid solubility and ranges from ~70% for alprazolam to nearly 99% for diazepam. The concentration in the cerebrospinal fluid is approximately equal to the concentration of free drug in plasma. The plasma concentrations of most benzodiazepines exhibit patterns that are consistent with 2-compartment models, but 3-compartment models appear to be more appropriate for the compounds with the highest lipid solubility. Accordingly, there is rapid uptake of benzodiazepines into the brain and other highly perfused organs after intravenous administration (or oral administration of a rapidly absorbed compound); rapid uptake is followed by a phase of redistribution into tissues that are less well perfused, especially muscle and fat. Redistribution is most rapid for drugs with the highest lipid solubility. The kinetics of redistribution of diazepam and other lipophilic benzodiazepines are complicated by enterohepatic circulation. The volumes of distribution of the benzodiazepines are large and in many cases are increased in elderly patients. These drugs cross the placental barrier and are secreted into breast milk.
Benzodiazepines are metabolized extensively by hepatic CYPs, particularly CYPs 3A4 and 2C19. Some benzodiazepines, such as oxazepam, are conjugated directly and are not metabolized by these enzymes. Erythromycin, clarithromycin, ritonavir, itraconazole, ketoconazole, nefazodone, and grapefruit juice are inhibitors of CYP3A4 (see Chapter 6) and can affect the metabolism of benzodiazepines. Because active metabolites of some benzodiazepines are biotransformed more slowly than are the parent compounds, the duration of action of many benzodiazepines bears little relationship to the t1/2 of elimination of the parent drug that was administered, as noted above for flurazepam. Conversely, the rate of biotransformation of agents that are inactivated by the initial reaction is an important determinant of their duration of action; these agents include oxazepam, lorazepam, temazepam, triazolam, and midazolam.
Because the benzodiazepines apparently do not significantly induce the synthesis of hepatic CYPs, their chronic administration usually does not result in the accelerated metabolism of other substances or of the benzodiazepines. Cimetidine and oral contraceptives inhibit N-dealkylation and 3-hydroxylation of benzodiazepines. Ethanol, isoniazid, and phenytoin are less effective in this regard. These reactions usually are reduced to a greater extent in elderly patients and in patients with chronic liver disease than are those involving conjugation.
PHARMACOKINETICS AND THE IDEAL HYPNOTIC.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


