Hyperlipidemia
KEY CONCEPTS
![]() Hypercholesterolemia, elevated low-density lipoprotein, and low high-density lipoprotein (HDL) are unequivocally linked to increased risk for coronary heart disease (CHD) and cerebrovascular morbidity and mortality; low-density lipoprotein (LDL) is the primary target.
Hypercholesterolemia, elevated low-density lipoprotein, and low high-density lipoprotein (HDL) are unequivocally linked to increased risk for coronary heart disease (CHD) and cerebrovascular morbidity and mortality; low-density lipoprotein (LDL) is the primary target.
![]() Multiple genetic abnormalities and environmental factors are involved in clinical lipid abnormalities and routinely used clinical laboratory measurements do not define the underlying abnormalities.
Multiple genetic abnormalities and environmental factors are involved in clinical lipid abnormalities and routinely used clinical laboratory measurements do not define the underlying abnormalities.
![]() Initial therapy for any lipoprotein disorder is therapeutic lifestyle changes with restricted intake of total and saturated fat and cholesterol and a modest increase in polyunsaturated fat intake along with a program of regular exercise and weight reduction if needed.
Initial therapy for any lipoprotein disorder is therapeutic lifestyle changes with restricted intake of total and saturated fat and cholesterol and a modest increase in polyunsaturated fat intake along with a program of regular exercise and weight reduction if needed.
![]() If therapy is insufficient after therapeutic lifestyle changes, lipid-lowering agents should be chosen based on the specific lipoprotein disorder presentation and the severity of the lipid abnormality.
If therapy is insufficient after therapeutic lifestyle changes, lipid-lowering agents should be chosen based on the specific lipoprotein disorder presentation and the severity of the lipid abnormality.
![]() Considering compliance, adverse effects, and effectiveness, statins are the drugs of choice for patients with hypercholesterolemia because they are the most potent form of monotherapy and are cost-effective in patients with known coronary artery disease (CAD) or multiple risk factors and in high-risk primary prevention patients.
Considering compliance, adverse effects, and effectiveness, statins are the drugs of choice for patients with hypercholesterolemia because they are the most potent form of monotherapy and are cost-effective in patients with known coronary artery disease (CAD) or multiple risk factors and in high-risk primary prevention patients.
![]() Patients not responding to statin monotherapy may be treated with combination therapy for hypercholesterolemia, but should be monitored closely because of an increased risk for adverse effects and drug interactions.
Patients not responding to statin monotherapy may be treated with combination therapy for hypercholesterolemia, but should be monitored closely because of an increased risk for adverse effects and drug interactions.
![]() Hypertriglyceridemia usually responds well to niacin, gemfibrozil, and fenofibrate; high-dose niacin should be used cautiously in diabetics because of worsening glycemic control. Statins lower triglycerides to a variable extent depending on baseline triglyceride concentration and statin potency.
Hypertriglyceridemia usually responds well to niacin, gemfibrozil, and fenofibrate; high-dose niacin should be used cautiously in diabetics because of worsening glycemic control. Statins lower triglycerides to a variable extent depending on baseline triglyceride concentration and statin potency.
![]() Low HDL-C is addressed with lifestyle modifications such as smoking cessation and increased exercise; niacin and gemfibrozil and fenofibrate can significantly increase HDL-C as well.
Low HDL-C is addressed with lifestyle modifications such as smoking cessation and increased exercise; niacin and gemfibrozil and fenofibrate can significantly increase HDL-C as well.
![]() Lipid-lowering therapy is generally considered to be cost-effective, particularly in secondary intervention and high-risk patients.
Lipid-lowering therapy is generally considered to be cost-effective, particularly in secondary intervention and high-risk patients.
![]() Reductions in elevated total cholesterol and LDL-C reduce CHD mortality and total mortality; increasing HDL may reduce CHD events as well. Aggressive treatment of hypercholesterolemia results in fewer patients progressing to myocardial infarction, angina, and stroke, and reduces the need for interventions such as coronary artery bypass graft and percutaneous transluminal coronary angioplasty.
Reductions in elevated total cholesterol and LDL-C reduce CHD mortality and total mortality; increasing HDL may reduce CHD events as well. Aggressive treatment of hypercholesterolemia results in fewer patients progressing to myocardial infarction, angina, and stroke, and reduces the need for interventions such as coronary artery bypass graft and percutaneous transluminal coronary angioplasty.
![]() Lomitapide and mipomersen have been recently approved for the treatment of homozygous familial hypercholesterolemia. Both have novel mechanisms of action to lower total and LDL cholesterol.
Lomitapide and mipomersen have been recently approved for the treatment of homozygous familial hypercholesterolemia. Both have novel mechanisms of action to lower total and LDL cholesterol.
Cholesterol, triglycerides, and phospholipids are the major lipids in the body and they are transported as complexes of lipid and proteins known as lipoproteins. Plasma lipoproteins are spherical particles with surfaces that consist largely of phospholipid, free cholesterol, and protein, and cores that consist mostly of triglyceride and cholesterol ester (Fig. 11-1). The three major classes of lipoproteins found in serum are low-density lipoproteins (LDL), high-density lipoproteins (HDL), and very-low-density lipoproteins (VLDL). VLDL is carried in the circulation as triglyceride and can be estimated by dividing the triglyceride concentration by 5 if the triglyceride concentration is below 250 mg/dL (2.83 mmol/L). Intermediate-density lipoprotein (IDL) resides between VLDL and LDL and is included in the LDL measurement in routine clinical measurement. Abnormalities of plasma lipoproteins can result in a predisposition to coronary, cerebrovascular, and peripheral vascular arterial disease and constitute one of the major risk factors for coronary heart disease (CHD). Accumulating evidence over the past decades had linked elevated total and LDL cholesterol and reduced HDL to the development of CHD. Premature coronary atherosclerosis, leading to the manifestations of ischemic heart disease (IHD; see Chap. 6), is the most common and significant consequence of dyslipidemia. The National Cholesterol Education Program (NCEP) Adult Treatment Panel III (ATP III) published its third report summarizing these data and giving recommendations for the management of hypercholesterolemia in adults.1,2 This report and the later update modify earlier recommendations and provide a new way of risk stratifying patients based on multiple risk factors, the presence of diabetes, and the metabolic syndrome. The American Heart Association (AHA) also provides guidelines for primary and secondary prevention of CHD.3–5
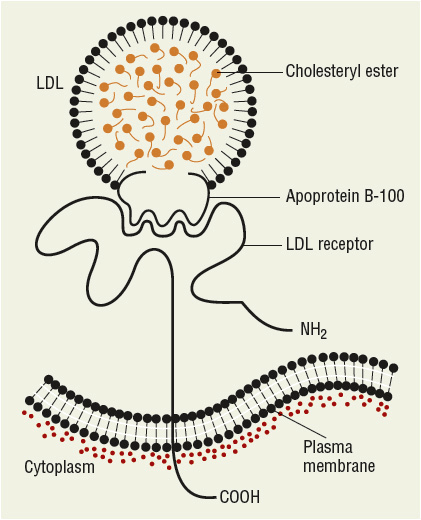
FIGURE 11-1 Diagrammatic representation of the structure of lowdensity lipoprotein (LDL), the LDL receptor, and the binding of LDL to the receptor via apolipoprotein B-100. (From Ganong WF. Review of Medical Physiology, 22nd ed. New York: McGraw-Hill, 2005:303.)
Total cholesterol and LDL-C increase throughout life in men and women, representing an atherogenic pattern characteristic of Westernized society diets.6 Based on estimates from the AHA, 43.4% or 98.9 million American adults over age 20 years have total cholesterol levels of 200 mg/dL (5.17 mmol/L) or higher.7 More than half of individuals at borderline high risk remain unaware that they have hypercholesterolemia and fewer than half of highest-risk persons (those with symptomatic CHD) are receiving lipid-lowering treatment. About one-third of treated patients are achieving their LDL goal; fewer than 20% of CHD patients are at their LDL goal.8,9 Changes in the NCEP guidelines have increased the number of persons eligible for therapeutic lifestyle changes (TLCs) or lipid-lowering therapy by millions. NCEP estimates that only 26% of patients have an optimal LDL-C (<100 mg/dL [<2.59 mmol/L]) and that large numbers of patients are either untreated or undertreated.1 Unfortunately, those patients at highest risk are less likely to be treated to desirable levels of LDL.10 Although these numbers seem staggering in their enormity, substantial progress has been made, and the number of Americans with a desirable blood cholesterol level (<200 mg/dL [<5.17 mmol/L]) has risen to 49% from 45% from the earlier survey (1976 to 1980), while the average total cholesterol in this country has fallen from 220 mg/dL (5.69 mmol/L) in 1960 to 195 mg/dL for men and 201 mg/dL for women.7 Patients who are at risk but who have not yet experienced their first cardiovascular or cerebrovascular event (e.g., myocardial infarction [MI]) are termed primary prevention, whereas those with manifest vascular disease are termed secondary intervention.
![]() Data from the Framingham study and from other studies demonstrate that the risk for developing cardiovascular disease is related to the degree of total cholesterol and LDL elevation in a graded, continuous fashion.11 Hypercholesterolemia is additive to the other nonlipid risk factors for CHD, including cigarette smoking, hypertension, diabetes, low HDL levels, and electrocardiographic abnormalities. The presence of established CHD or prior MI increases the risk of MI five to seven times that seen in men or women without CHD, and LDL is a significant predictor of subsequent morbidity and mortality. About 50% of all MIs and at least 70% of CHD deaths occur in patients with known CHD, and these patients should therefore be a target for screening, identification, and treatment. Unfortunately, the identification of patients at high risk because of hypercholesterolemia or other lipid disorders is too frequently overlooked, because blood lipid levels are not always evaluated in this population even after an event such as MI.
Data from the Framingham study and from other studies demonstrate that the risk for developing cardiovascular disease is related to the degree of total cholesterol and LDL elevation in a graded, continuous fashion.11 Hypercholesterolemia is additive to the other nonlipid risk factors for CHD, including cigarette smoking, hypertension, diabetes, low HDL levels, and electrocardiographic abnormalities. The presence of established CHD or prior MI increases the risk of MI five to seven times that seen in men or women without CHD, and LDL is a significant predictor of subsequent morbidity and mortality. About 50% of all MIs and at least 70% of CHD deaths occur in patients with known CHD, and these patients should therefore be a target for screening, identification, and treatment. Unfortunately, the identification of patients at high risk because of hypercholesterolemia or other lipid disorders is too frequently overlooked, because blood lipid levels are not always evaluated in this population even after an event such as MI.
A comparison of the United States to other countries shows similar relationships between total cholesterol and LDL, and an inverse relationship with HDL to coronary artery disease (CAD) mortality.11 On a positive note, the U.S. mortality rate is midway among the countries studied, and this country has had the greatest decline in CAD mortality (35% to 40%) in men and women over the past 10 years as compared with other countries. A decline in the prevalence of hypercholesterolemia in certain segments of the U.S. population parallels these trends in mortality.1 LDL and the ratio of LDL to HDL have also been used to assess risk, but their use adds little information to total cholesterol alone unless HDL is abnormally high or low. McQueen et al. found that the ratio of apolipoprotein (Apo) B to ApoA-I was more predictive and consistent across gender and ethnic groups.12 HDL transports cholesterol from lipid-laden foam cells to the liver. HDL has been shown to be protective for the occurrence of CHD, and an inverse relationship exists between CHD and HDL levels.13 Recent clinical trials attempting to raise HDL have failed to demonstrate clinically meaningful reductions in cardiovascular end points challenging the importance of increasing HDL fractions and ApoA-I.14
VLDL, the major lipoprotein associated with triglycerides, is enriched with cholesterol esters, and is smaller, denser, and more atherogenic than less dense VLDL. Routine measurement of triglycerides cannot distinguish between the types of VLDL present in plasma. Elevation of triglyceride-rich lipoproteins is associated with low HDL, and this ratio predicts increased risk. The 8-year followup of the Copenhagen male study found a clear gradient of risk of IHD with increasing triglyceride levels within each level of HDL cholesterol. When compared with the lowest tertile of triglyceride concentrations, the highest tertile had 2.2 relative risk for IHD and the relationship extended across all concentrations of HDL.15 The Helsinki Heart Study shows that hypertriglyceridemia and low HDL are associated with obesity (body mass index [BMI] >26 kg/m2), smoking, sedentary lifestyle, blood pressure of ≥140/90 mm Hg, and blood glucose above 79 mg/dL (4.4 mmol/L), and that the benefit of gemfibrozil (risk reduction 68%, P <0.03) was largely confined to overweight subjects.16 Hypertriglyceridemia in certain instances—for example, diabetes mellitus, nephrotic syndrome, and chronic renal disease, and perhaps in women—is associated with increased cardiovascular risk. This is thought to be a consequence of the presence of atherogenic lipoproteins and of hypertriglyceridemia being a marker for them, as triglycerides are usually not independently predictive for CHD.17
LIPOPROTEIN METABOLISM AND TRANSPORT
Cholesterol and triglycerides, as the major plasma lipids, are essential substrates for cell membrane formation and hormone synthesis, and provide a source of free fatty acids.18 Dyslipidemia may be defined as an elevation in total cholesterol, elevation in LDL cholesterol, elevation in triglycerides or low HDL cholesterol concentration, or some combination of these abnormalities. Lipids, being water immiscible, are not present in free form in the plasma, but rather circulate as lipoproteins. Hyperlipoproteinemia describes an increased concentration of the lipoprotein macromolecules that transport lipids in the plasma. The density of plasma lipoproteins is determined by their relative content of protein and lipid. Density, composition, size, and electrophoretic mobility divide lipoproteins into four classes (Table 11-1).
TABLE 11-1 Composition of Lipoprotein Isolated from Normal Subjects
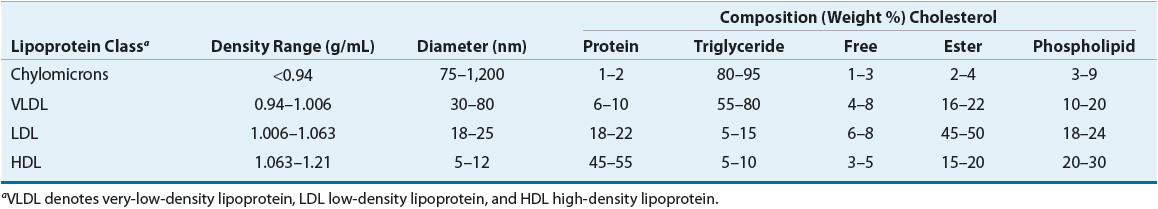
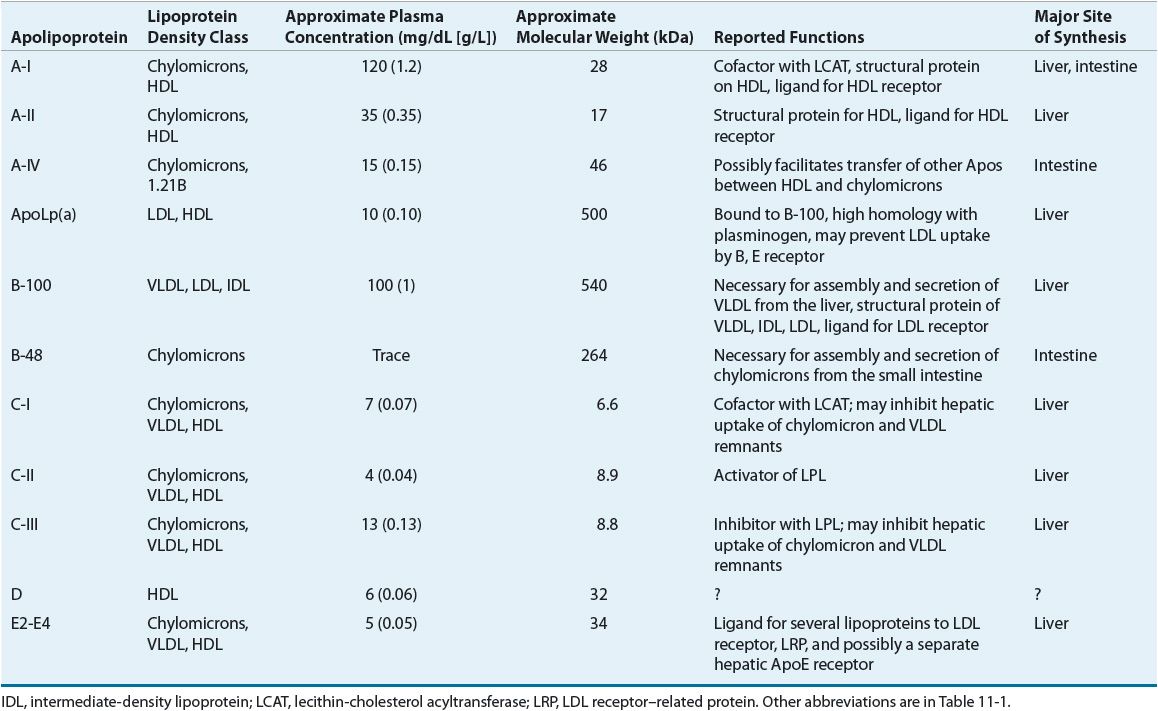
LDL has been further divided into LDL1, or IDL (density 1.006 to 1.019 g/mL), and LDL2 (1.019 to 1.063 g/mL). LDL2 is the major LDL component in plasma and it carries 60% to 70% of the total serum cholesterol. HDL has been subfractionated into HDL2 (density 1.063 to 1.125 g/mL) and HDL3 (1.125 to 1.21 g/mL). Fluctuations in HDL are usually caused by alterations in the levels of HDL2. HDL normally carries about 20% to 30% of the total cholesterol. VLDL has also been subdivided into three classes, and it carries about 10% to 15% of serum cholesterol and most of the triglycerides in the fasting state. VLDL is the precursor for LDL, and VLDL remnants may also be atherogenic. Table 11-2 shows the characteristics of the protein constituent of lipoproteins known as Apos. The structure of LDL, the LDL receptor (LDL-R), and the binding of the LDL to the receptor via ApoB-100 are shown in Figure 11-1.
TABLE 11-2 Characteristics and Functions of Apolipoproteins
Chylomicrons, large triglyceride-rich particles containing Apos B-48, B-100, and E, are formed from dietary fat solubilized by bile salts in intestinal mucosal cells. Chylomicrons are normally not present in the plasma after a fast of 12 to 14 hours and are catabolized by lipoprotein lipase (LPL), which is activated by ApoC-II and in the vascular endothelium and hepatic lipase to form chylomicron remnants. The remnants that contain ApoE (Fig. 11-2) are taken up by the “remnant receptor,” which may be an LDL-R-related protein, in the liver. Free cholesterol is liberated intracellularly after attachment to the remnant receptor. Chylomicrons also function to deliver dietary triglyceride to skeletal muscle and adipose tissue. During the catabolism of nascent chylomicrons to remnants, triglyceride is converted to free fatty acids and Apos A-I, A-II, A-IV (free in plasma), C-I, C-II, and C-III, and phospholipids are transferred to HDL. Apos E and C-II are transferred to chylomicrons from HDL and eventually back through these metabolic events. Hepatic VLDL synthesis is regulated in part by diet and hormones, and is inhibited by uptake of chylomicron remnants in the liver. VLDL is secreted from the liver and serially converted via LPL to IDL, and, finally, to LDL. VLDL receptors are found in adipose tissue and muscle, and bear close homology to the structure of LDL-Rs.
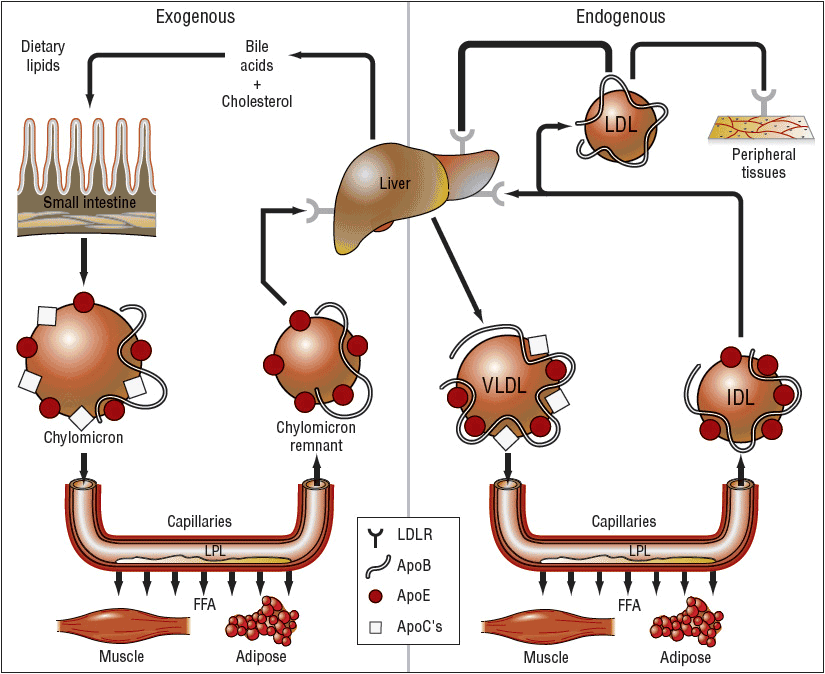
FIGURE 11-2 Simplified diagram of lipoprotein systems for transporting lipids in humans. In the exogenous system, chylomicrons rich in triglycerides of dietary origin are converted to chylomicron remnants rich in cholesteryl esters by the action of lipoprotein lipase (LPL). In the endogenous system, very-low-density lipoproteins (VLDL) rich in triglycerides are secreted by the liver and converted to intermediate-density lipoproteins (LDL) and then to low-density lipoproteins (LDL) rich in cholesteryl esters. Some of the LDLs enter the subendothelial space of arteries, are oxidized, and then are taken up by macrophages, which become foam cells. The letters on the chylomicrons, chylomicron remnants, VLDL, IDL, and LDL identify the primary apoproteins (ApoB, ApoC, ApoE) found in them. (LDLR, low-density lipoprotein receptor.) (From Kasper DL, Braunwald E, Fauci AS, et al., eds. Harrison’s Principles of Internal Medicine, 16th ed. New York, McGraw-Hill, 2005, p. 2289.)
LDL, the major cholesterol transport lipoprotein and having virtually only ApoB-100, is mostly derived from VLDL catabolism and cellular synthesis. When fasting and on low-fat intake in normal subjects, most cholesterol is synthesized and used in the extrahepatic organs, while most of the cholesterol carried by LDL is taken up by the liver for catabolism. In patients with homozygous familial hypercholesterolemia, enhanced synthesis of LDL may occur, because LDL clearance is reduced as a consequence of the lack of LDL-Rs. LDL is catabolized through interaction of cell surface receptors found on liver, adrenal, and peripheral cells (including fibroblasts and smooth muscle cells). These cells recognize ApoB-100 on LDL, and after binding to a receptor on the cell membrane, LDL is internalized and degraded. In the normal fasting state, approximately 70% of LDL is cleared through receptor-dependent mechanism, although this is highly dependent on the availability and type of saturated and monounsaturated or polyunsaturated fat from dietary sources. Ingestion of cholesterol and saturated fatty acids such as C12:0, C14:0, and C16:0 is associated with reduction in LDL-R activity, increased LDL production rate, and elevation in LDL plasma concentration. Receptor-independent mechanisms are also involved to a lesser extent in the catabolism of LDL, and these receptors are present in many tissues but are most active in animals in the adrenals and ovary. Increased intracellular cholesterol resulting from LDL catabolism inhibits the activity of 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMG-CoA reductase), the rate-limiting enzyme for intracellular cholesterol biosynthesis (Fig. 11-3). Additional consequences of increased intracellular cholesterol include reduced synthesis of LDL-R, which limits subsequent cholesterol uptake from the plasma, and accelerated activity of acyl-coenzyme A-to-cholesterol acyltransferase (ACAT) to facilitate cholesterol storage within cells. LDL cholesterol may also be excreted into bile and become part of the enterohepatic pool or may be lost in the stool. Lp(a) is a cholesterol-rich lipoprotein similar to LDL in composition and density and with close homology to fibrinogen; it is reported to be an important independent risk factor for the development of premature cardiovascular disease.
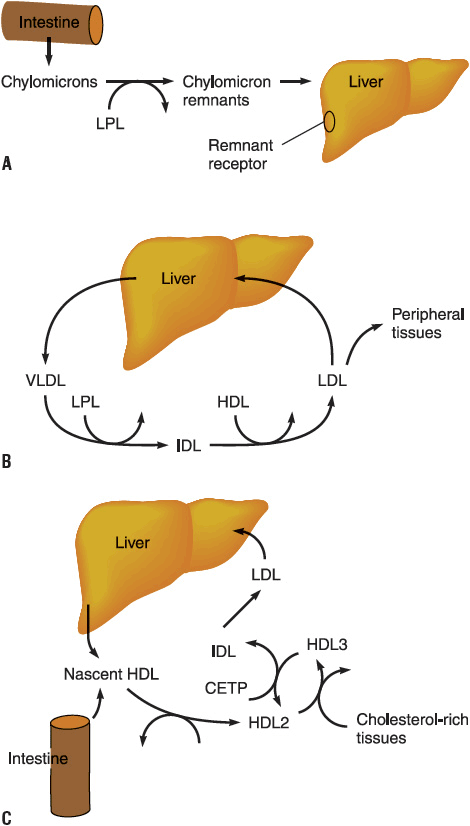
FIGURE 11-3 Biosynthetic pathway for cholesterol. The rate-limiting enzyme in this pathway is 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMG-CoA reductase). (CETP, cholesterol ester transfer protein; HDL, high-density lipoprotein; IDL, intermediate-density lipoprotein; LDL, low-density lipoprotein; LPL, lipoprotein lipase; VLDL, very-low-density lipoprotein.) A. Exogenous pathway; B. Endogenous pathway; C. Reverse cholesterol transport. (Modified from Breslow JL. Genetic basis of lipoprotein disorders. J Clin Invest 1989;84:373.)
Nascent HDL is derived from liver and gut synthesis primarily in the form of ApoA-I phospholipid disks.13 Esterification of free cholesterol in nascent HDL and from peripheral tissues to cholesteryl esters by lecithin-cholesterol acyltransferase (LCAT) results in the production of HDL3. Further addition of tissue cholesterol to HDL3 results in the formation of HDL2. HDL2 can also be formed from remodeling of chylomicrons and VLDL catabolism. It may be converted back to HDL3 by the action of hepatic lipase and by the transfer of cholesteryl esters to the liver, LDL, and VLDL. ApoA-I production is increased by estrogens, leading to higher HDL levels in women and in individuals receiving estrogen. Transfer of excess cholesterol from peripheral tissues by HDL is called reverse cholesterol transport. Putative HDL receptors in peripheral cells facilitate the uptake of cholesterol by HDL, which transfers cholesterol to either VLDL and LDL or the liver for secretion into bile or conversion into bile acids. These processes serve to rid peripheral tissue (e.g., coronary arteries) of excessive amounts of cholesterol, and account for some of the protective effects noted with increasing HDL in women and other factors that elevate HDL levels. Variants of the cholesterol ester transfer protein (CETP) have been demonstrated in humans, and the B1B1 genotype is associated with lower HDL and progression of coronary atherosclerosis. Inhibition of CETP leads to elevations in HDL; unfortunately when CETP inhibitors were tested in clinical trials, they did not induce regression of atherosclerotic plaque and were associated with higher blood pressure and CHD events.19–22 The effect of CETP inhibition on blood pressure and HDL is disconcordant with some of these agents.23
The “response-to-injury” hypothesis states that risk factors such as oxidized LDL, mechanical injury to the endothelium (e.g., percutaneous transluminal angioplasty), excessive homocysteine, immunologic attack, or infection-induced (e.g., Chlamydia, herpes simplex virus-1) changes in endothelial and intimal function lead to endothelial dysfunction and a series of cellular interactions that culminate in atherosclerosis. C-reactive protein (CRP) is an acute phase reactant and a marker for inflammation; it may be useful in identifying patients at risk for developing CAD.24 The transcription factor Kruppel-like factor 2 (KLF2) may be induced by statins in liver sinusoidal endothelial cells (SEC), orchestrating an efficient vasoprotective response. Upregulation of hepatic endothelial KLF2-derived transcriptional programs by statins confers vasoprotection and stellate cell deactivation, reinforcing the therapeutic potential of these drugs for liver diseases that course with endothelial dysfunction.
The eventual outcomes of this atherogenic cascade are clinical events such as angina, MI, arrhythmias, stroke, peripheral arterial disease, abdominal aortic aneurysm, and sudden death. Atherosclerotic lesions are thought to arise from transport and retention of plasma LDL cholesterol through the endothelial cell layer into the extracellular matrix of the subendothelial space. Once in the artery wall, LDL is chemically modified through oxidation and nonenzymatic glycation. Mildly oxidized LDL then recruits monocytes into the artery wall, which become transformed into macrophages. Macrophages have tremendous potential for accelerating LDL oxidation and ApoB accumulation, and altering the receptor-mediated uptake of LDL into the artery wall from the usual LDL-R to a “scavenger receptor” not regulated by cell content of cholesterol. Oxidized LDL increases plasminogen inhibitor levels (promotion of coagulation), induces the expression of endothelin (vasoconstrictive substance), inhibits the expression of nitric oxide (a vasodilator and platelet inhibitor), and is toxic to macrophages if highly oxidized. As oxidation of biologically active lipids proceeds, other lipids such as lysophosphatidylcholine, hydroperoxides, aldehydic breakdown products of fatty acids, and oxysterol are formed, which continue the reaction within the tissue. These events lead to a massive accumulation of cholesterol. The cholesterol-laden macrophages become foam cells; foam cells are the earliest recognized cells of the arterial fatty streak.
Oxidized LDL provokes an inflammatory response, which is mediated by a number of chemoattractants and cytokines. Examples of each that appear to be involved at different stages of lesion development include monocyte chemoattractant protein 1 (MCP-1), monocyte colony-stimulating factor (M-CSF), gro, vascular cell adhesion molecule (VCAM-1), E-selectin (ELAM-1), intercellular adhesion molecule (ICAM-1), platelet-derived growth factor (PDGF), vascular endothelial growth factor (VEGF), transforming growth factors (TGF-α and TGF-β), interleukin-1 (IL-1) and interleukin-6 (IL-6), and the ratio of interleukin-10 (IL-10) and interleukin-12 (IL-12). It appears that some of these factors (e.g., MCP-1 and M-CSF) participate early in the process of monocyte–macrophage attachment and transmigration across the endothelium, whereas others (PDGF and VCAM-1) promote later lesion growth.25 The extent of oxidation and the inflammatory response is under genetic control of a major gene termed Ath-1 based on murine model studies. The process of aging may lead to lipoproteins that are more susceptible to oxidation and have longer resident time in the vascular compartment. Two proteins associated with HDL—ApoJ and paraoxonase (PON)—appear to play an important role to minimize the oxidation of LDL-C.26 Increased recognition of the role of these growth-regulatory molecules provides the possibility of future directions for antagonists to regulatory molecules such as PDGF, TGF-β, and the interleukins. Repeated injury and repair within an atherosclerotic plaque eventually lead to a fibrous cap protecting the underlying core of lipids, collagen, calcium, and inflammatory cells such as T lymphocytes. Maintenance of the fibrous plaque is critical to prevent plaque rupture and subsequent coronary thrombosis.27 An imbalance between plaque synthesis and degradation may lead to a weakened or vulnerable plaque prone to rupture. The fibrous cap may become weakened through decreased synthesis of the extracellular matrix or increased degradation of the matrix. The cytokine interferon-γ, produced by T lymphocytes, inhibits the ability of smooth muscle cells to synthesize collagen, a structurally important component of the fibrous cap. A family of enzymes known as matrix metalloproteinases can degrade all major constituents of the vascular extracellular matrix: collagen, elastin, and proteoglycans.28
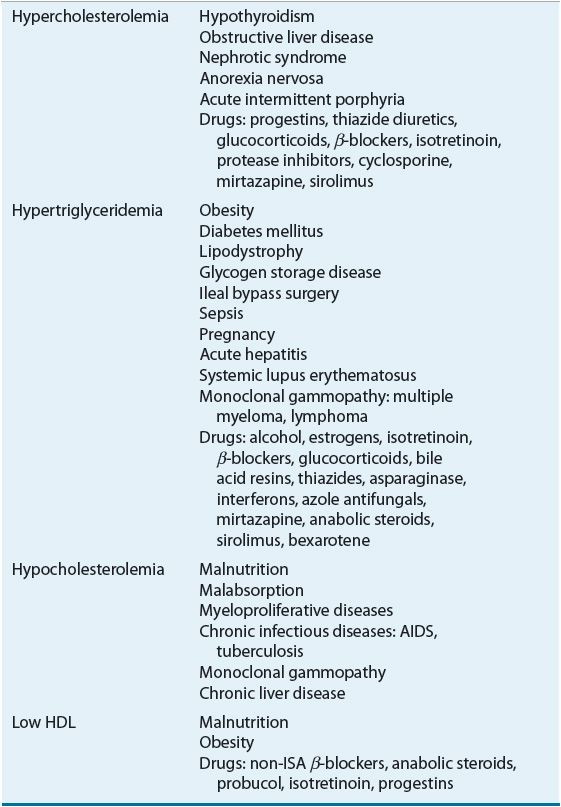
![]() Lipoprotein disorders are classified into six categories, which are commonly used for phenotypical description of dyslipidemia (Table 11-3). Specific genetic defects with disrupted protein, cell, and organ function give rise to several disorders within each family of lipoproteins (Table 11-4). In other words, an elevated cholesterol level does not necessarily equate with familial hypercholesterolemia or type IIa, as cholesterol may also be elevated in other lipoprotein disorders and the lipoprotein pattern does not describe the underlying genetic defect. The preceding discussion has focused on primary or genetic dyslipoproteinemia; it should be remembered that secondary forms exist and that several drugs may also elevate lipid levels (Table 11-5). These secondary forms of hyperlipidemia should be initially managed by correcting the underlying abnormality, including modification of drug therapy when appropriate.
Lipoprotein disorders are classified into six categories, which are commonly used for phenotypical description of dyslipidemia (Table 11-3). Specific genetic defects with disrupted protein, cell, and organ function give rise to several disorders within each family of lipoproteins (Table 11-4). In other words, an elevated cholesterol level does not necessarily equate with familial hypercholesterolemia or type IIa, as cholesterol may also be elevated in other lipoprotein disorders and the lipoprotein pattern does not describe the underlying genetic defect. The preceding discussion has focused on primary or genetic dyslipoproteinemia; it should be remembered that secondary forms exist and that several drugs may also elevate lipid levels (Table 11-5). These secondary forms of hyperlipidemia should be initially managed by correcting the underlying abnormality, including modification of drug therapy when appropriate.
TABLE 11-3 Fredrickson-Levy-Lees Classification of Hyperlipoproteinemia
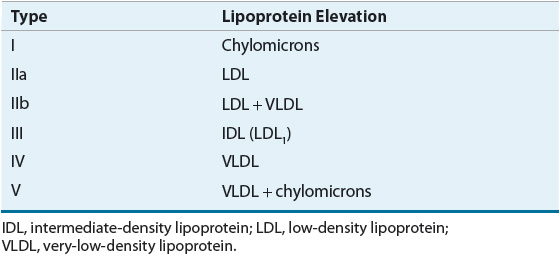
TABLE 11-4 Lipoprotein Disorders
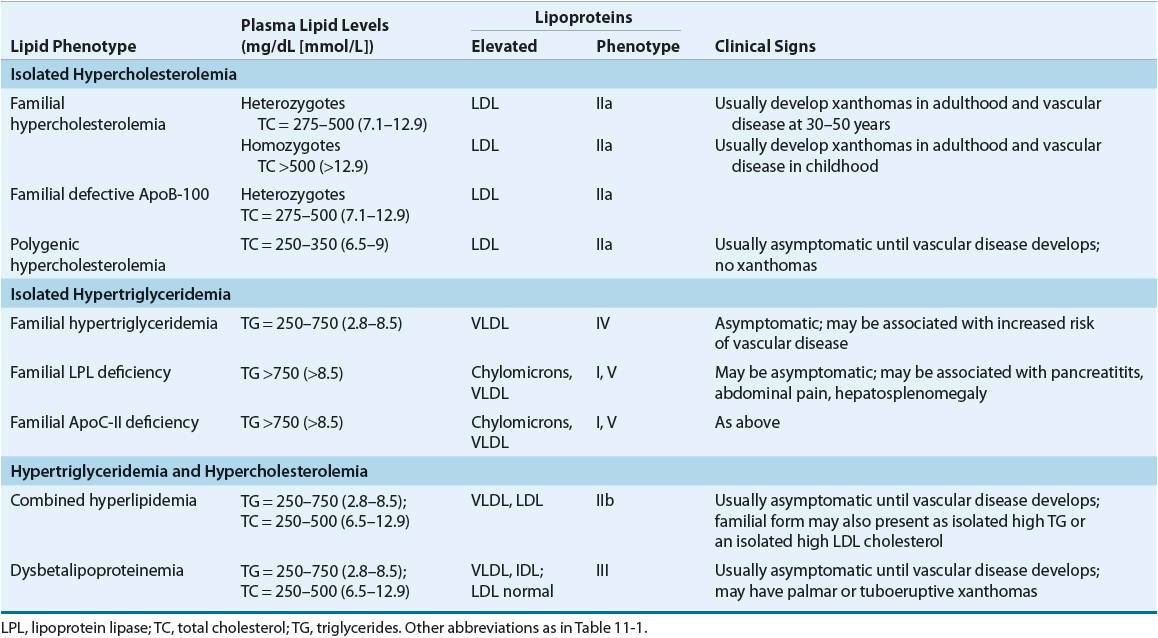
TABLE 11-5 Secondary Causes of Lipoprotein Abnormalities
Familial hypercholesterolemia is characterized by (a) a selective elevation in the plasma level of LDL; (b) deposition of LDL-derived cholesterol in tendons (xanthomas) and arteries (atheromas); and (c) inheritance as an autosomal dominant trait with homozygotes more severely affected than heterozygotes. Homozygotes (prevalence 1 in 1,000,000) have severe hypercholesterolemia (650 to 1,000 mg/dL [16.8 to 25.9 mmol/L]), with the early appearance of cutaneous xanthomas and fatal CHD generally before the age of 20. The primary defect in familial hypercholesterolemia is the inability to bind LDL to the LDL-R or, rarely, a defect of internalizing the LDL-R complex into the cell after normal binding. Homozygotes have essentially no functional LDL-Rs. This leads to lack of LDL degradation by cells and unregulated biosynthesis of cholesterol, with total cholesterol and LDL-C being inversely proportional to the deficit in LDL-Rs. Heterozygotes have only about one half of the normal number of LDL-Rs, total cholesterol levels in the range of 300 to 600 mg/dL (7.76 to 15.52 mmol/L), and cardiovascular events beginning in the third and fourth decades of life.
Familial LPL deficiency is a rare, autosomal recessive trait characterized by a massive accumulation of chylomicrons and corresponding increase in plasma triglycerides or a type I lipoprotein pattern. VLDL concentration is normal. The presenting manifestations include repeated attacks of pancreatitis and abdominal pain, eruptive cutaneous xanthomatosis, and hepatosplenomegaly beginning in childhood. Symptom severity is proportional to dietary fat intake, and consequently to the elevation of chylomicrons. LPL is normally released from vascular endothelium or by heparin and hydrolyzes chylomicrons and VLDL (see Fig. 11-2). Diagnosis is based on low or absent enzyme activity with normal human plasma or ApoC-II, a cofactor of the enzyme. Accelerated atherosclerosis is not associated with this disease. Abdominal pain, pancreatitis, eruptive xanthomas, and peripheral polyneuropathy characterize type V (VLDL and chylomicrons). Symptoms may occur in childhood, but usually the disorder is expressed at a later age. The risk of atherosclerosis is increased with this disorder. These patients are commonly obese, hyperuricemic, and diabetic, and alcohol intake, exogenous estrogens, and renal insufficiency tend to be exacerbating factors.
CLINICAL PRESENTATION
Patients with familial type III hyperlipoproteinemia (also called dysbetalipoproteinemia, broadband, or β-VLDL) develop the following clinical features after 20 years of age: xanthoma striata palmaris (yellow discolorations of the palmar and digital creases), tuberous or tuberoeruptive xanthomas (bulbous cutaneous xanthomas), and severe atherosclerosis involving the coronary arteries, internal carotids, and abdominal aorta. A defective structure of ApoE does not allow normal hepatic surface receptor binding of remnant particles derived from chylomicrons and VLDL (known as IDL); aggravating factors such as obesity, diabetes, or pregnancy may promote overproduction of ApoB–containing lipoproteins. Although homozygosity for the defective allele (E2/E2) is common (1 in 100), only 1 in 10,000 expresses the full-blown picture, and interaction with other genetic or environmental factors, or both, is needed to produce clinical disease.
Familial combined hyperlipidemia is characterized by elevations in total cholesterol, triglycerides, decreased HDL, increased ApoB, and small, dense LDL.29 It is associated with premature CHD and may be difficult to diagnose since the lipid levels do not consistently display the same pattern.
Type IV hyperlipoproteinemia is common and occurs in adulthood primarily in patients who are obese, diabetic, and hyperuricemic and do not have xanthomas. It may be secondary to alcohol ingestion and can be aggravated by stress, progestins, oral contraceptives, thiazides, or β-blockers. Two genetic patterns occur in type IV hyperlipoproteinemia: familial hypertriglyceridemia, which does not carry a great risk for premature CAD, and familial combined hyperlipidemia, which is associated with increased risk of cardiovascular disease.
Rare forms of lipoprotein disorders may include hypobetalipoproteinemia, abetalipoproteinemia, Tangier disease, LCAT deficiency (fish-eye disease), cerebrotendinous xanthomatosis (CTX), and sitosterolemia. Most of these rare lipoprotein disorders do not result in premature atherosclerosis, with the exceptions of familial LCAT deficiency, CTX, and sitosterolemia with xanthomatosis. Their treatment consists of dietary restriction of plant sterols (sitosterolemia with xanthomatosis), chenodeoxycholic acid (CTX), or, potentially, blood transfusion (LCAT deficiency).
PATIENT EVALUATION
A fasting lipoprotein profile including total cholesterol, LDL-C, HDL-C, and triglycerides should be measured in all adults 20 years of age or older at least once every 5 years.1 If the profile is obtained in the nonfasted state, only total cholesterol and HDL-C will be usable because LDL-C is usually a calculated value; if total cholesterol is ≥200 mg/dL (≥5.17 mmol/L), or if HDL-C is <40 mg/dL (<1.03 mmol/L), a followup fasting lipoprotein profile should be obtained. After a lipid abnormality is confirmed (Table 11-6), major components of the evaluation are the history (including age, gender, and, if female, menstrual and hormone replacement status), physical examination, and laboratory investigations. A complete history and physical examination should assess (a) presence or absence of cardiovascular risk factors (Table 11-7) or definite cardiovascular disease in the individual; (b) family history of premature cardiovascular disease or lipid disorders; (c) presence or absence of secondary causes of lipid abnormalities, including concurrent medications (see Table 11-5); and (d) presence or absence of xanthomas or abdominal pain, or history of pancreatitis, renal or liver disease, peripheral vascular disease, abdominal aortic aneurysm, or cerebral vascular disease (carotid bruits, stroke, or transient ischemic attack [TIA]). An important change in the ATP III guidelines is that diabetes mellitus is regarded as a CHD risk equivalent.1 The presence of diabetes in patients without known CHD is associated with the same level of risk as in patients without diabetes but having confirmed CHD.30,31 ATP III identifies four categories of risk that modify the goals and modalities of LDL-lowering therapy (Table 11-8).2 The highest category is known CHD or CHD risk equivalents, which is defined as the risk for major coronary events equal to or greater than established CHD, that is, >20% per 10 years (2% per year). The next category is moderately high risk consisting of patients with multiple (2+) risk factors in which 10-year risk for CHD is 10% to 20%. Moderate risk is defined as ≥2 risk factors and a 10-year risk of ≤10%. The lowest risk category is persons with zero to one risk factor. Risk is estimated from Framingham risk scores.32 Risk is estimated based on the patient’s age, LDL-C or total cholesterol level, blood pressure, the presence of diabetes, and smoking status (Table 11-7). This approach for a single patient is referred to as case finding or patient-based approach, whereas large-scale screening and recommendations for the general populace, healthcare providers, and the food industry are called a population-based approach.
TABLE 11-6 Classification of Total, LDL, and HDL Cholesterol, and Triglycerides
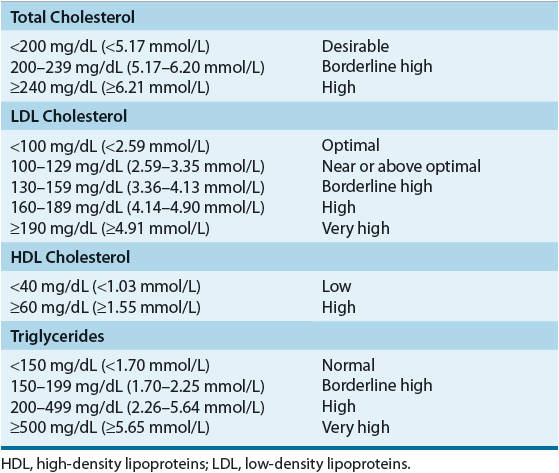
TABLE 11-7 Major Risk Factors (Exclusive of LDL Cholesterol) that Modify LDL Goalsa

TABLE 11-8 LDL Cholesterol Goals and Cut Points for Therapeutic Lifestyle Changes (TLCs) and Drug Therapy in Different Risk Categoriesa

Measurement of plasma cholesterol (which is about 3% lower than serum determinations), triglyceride, and HDL-C levels after a 12-hour or longer fast is important, as triglycerides may be elevated in nonfasted individuals; total cholesterol is only modestly affected by fasting. Analytic and biologic variability can have a major impact on the measurement and interpretation of cholesterol (or any other laboratory test). Analytic variability can be minimized through the use of adequate quality control procedures, including internal training, routine calibration and monitoring, and external proficiency testing. Even with these measures, the coefficient of variability in the best procedures can acceptably be up to 5%, and when combined with average biologic variability, total variability may be as high as about 22%. Analytic variability with desktop equipment generally is greater in the finger-stick capillary blood methods, usually yielding measurements less than those from a clinical laboratory, and this technology should be considered for use only as a screening method. Reliance on desktop methods can result in misclassification of 7% to 14% of patients if capillary blood is used. Two determinations, 1 to 8 weeks apart, with the patient on a stable diet and weight, and in the absence of acute illness, are recommended to minimize variability and to obtain a reliable baseline.1 If the total cholesterol is greater than 200 mg/dL (5.17 mmol/L), a second determination is recommended, and if the values are more than 30 mg/dL (0.78 mmol/L) apart, the average of three values should be used. Familiarity with the method and quality control procedures employed by local laboratories is essential for interpretation of reported values. If the physical examination and history are insufficient to diagnose a familial disorder, then agarose-gel lipoprotein electrophoresis is useful to determine which class of lipoproteins is affected. If the triglyceride levels are below 400 mg/dL (4.52 mmol/L) and neither type III hyperlipidemia nor chylomicrons are detected by electrophoresis, then one can calculate VLDL and LDL concentrations: VLDL = triglyceride/5; LDL = total cholesterol – (VLDL – HDL).
Because total cholesterol is composed of cholesterol derived from LDL, VLDL, and HDL, determination of HDL is useful when total plasma cholesterol is elevated. HDL may be elevated by moderate alcohol ingestion (less than two drinks per day), physical exercise, smoking cessation, weight loss, oral contraceptives, phenytoin, and terbutaline. Smoking, obesity, a sedentary lifestyle, and drugs such as β-blockers lower HDL. Only exercise and smoking cessation could be recommended as interventions for low HDL concentrations. Niacin and gemfibrozil also increase HDL concentrations.
The range of lipid concentrations represents a population mean plus or minus two standard deviations and does not define the risk of disease. Reference values for plasma total, LDL, and HDL cholesterol concentrations for men and women, as well as various ethnic groups, are available from the NHANES III.6 Cholesterol and triglycerides increase throughout life until about the fifth decade for men and the sixth decade for women. Past these ages, total cholesterol and LDL plateau and fall slightly. HDL tends to fall slightly with time and more rapidly after menopause in women. Institution of a population-based approach for cholesterol reduction should shift the entire curve to the left, and the potential reduction in cardiovascular mortality would be proportional to mean reductions at any cholesterol concentration.
Based on a careful review of the experimental pathologic, genetic, and epidemiologic evidence relating to the relationship between blood cholesterol levels and CHD, the ATP III of the NCEP recommends that a fasting lipoprotein profile and risk factor assessment be used in the initial classification of adults.1,33 If total cholesterol is less than 200 mg/dL (5.17 mmol/L), then the patient has a desirable blood cholesterol level (Table 11-6). Cholesterol levels between 200 and 239 mg/dL (5.17 and 6.18 mmol/L) are classified as borderline high blood cholesterol levels, and assessment of risk factors (Table 11-7) is needed to more clearly define disease risk. Blood cholesterol levels of 240 mg/dL (6.21 mmol/L) and above are classified as high blood cholesterol levels. Clinicians are awaiting the publication of ATP IV and it is anticipated that these categories of lipoproteins may be revised downward. If the total cholesterol is below 200 mg/dL (5.17 mmol/L) and the HDL is above 40 mg/dL (1.03 mmol/L), no further followup is recommended for patients without known CHD and who have fewer than two risk factors. In patients with evidence of CHD or other clinical atherosclerotic disease, the LDL goal is less than 100 mg/dL (2.59 mmol/L) and most patients will require diet and/or drug intervention. In patients with very high risk (known CHD and multiple risk factors) the LDL goal may be set at <70 mg/dL (<1.81 mmol/L) based on evidence from newer studies.33 Decisions regarding classification and management are based on the LDL-C levels as outlined in Table 11-8. An increasing number of persons have the metabolic syndrome that is characterized by abdominal obesity, atherogenic dyslipidemia (elevated triglycerides, small LDL particles, low HDL-C), raised blood pressure, insulin resistance (with or without glucose intolerance), and prothrombotic and proinflammatory states. ATP III recognizes the metabolic syndrome as a secondary target of risk reduction therapy after LDL-C has been addressed and if the metabolic syndrome is present, the patient is considered to have a CHD risk equivalent. Other lipid targets include non-HDL goals for patients with triglycerides >200 mg/dL (>2.26 mmol/L). Non-HDL is calculated by subtracting HDL from total cholesterol and the targets are 30 mg/dL (0.78 mmol/L) greater than LDL for each risk stratum. Non-HDL takes into consideration atherogenic particles such as remnant lipoproteins and IDL that are not measured in routine clinical laboratory testing.34 HDL raising has potential benefit, but no specific goals are set in the current guidelines and the evidence is modest to support aggressively increasing HDL levels.35
The Expert Panel on Children and Adolescents of the NCEP recommends screening in higher-risk children (positive family history or parental high blood cholesterol, ≥240 mg/dL [≥6.21 mmol/L]).36 The American Academy of Pediatrics categorizes total and LDL cholesterol into acceptable (<75th percentile; total cholesterol <170 mg/dL [<4.40 mmol/L]), borderline (75th to 95th percentile; total cholesterol 170 to 199 mg/dL [4.40 to 5.15 mmol/L], LDL cholesterol 110 to 129 mg/dL [2.84 to 3.34 mmol/L]), and elevated (>95th percentile; total cholesterol >200 mg/dL [>5.17 mmol/L], LDL cholesterol >130 mg/dL [>3.36 mmol/L]).36 The rationale, in part, for this approach is based on the recognition that atherosclerosis begins in the childhood and adolescent years as documented in the pathobiologic determinants of atherosclerosis in youth (PDAY) and the Bogalusa studies.37 Similarly, if children with high blood lipids or lipoprotein levels are identified, and the levels in the parents are unknown, the parents should be screened as well, as they are likely to be at high risk. Racial and gender differences do exist in the determination of lipoprotein fractions, and these factors should be considered in screening. Use of the serum cholesterol level alone may be of insufficient specificity or sensitivity, depending on the cut points used in screening, and other discretionary factors, such as hypertension, smoking, obesity, high-fat diet, and use of cholesterol-raising medication, may be needed to correctly identify children at risk. Presently, children over the age of 10 years are candidates for drug therapy if a trial of diet (6 months to 1 year) proves to be inadequate and LDL-C remains above 190 mg/dL (4.91 mmol/L), or above 160 mg/dL (4.14 mmol/L) if two or more risk factors or CHD are present in the child or adolescent, or if there is a history of premature CHD. In children with diabetes mellitus, pharmacologic treatment should be considered when LDL cholesterol is ≥130 mg/dL (≥3.36 mmol/L).36 The Dietary Intervention Study in Children (DISC) in pubertal children found that a fat-restricted diet modestly lowered LDL-C and maintained psychological well-being and dietary changes are acceptable to children.38,39 Although bile acid sequestrants have been the recommended drugs for this population, clinical trials demonstrate that statin therapy is effective and well tolerated in pediatric populations.40,41 The long-term consequences of drug therapy in this population are unknown. In special instances, familial hypercholesterolemia (particularly the homozygous form), or the existence of CHD or two or more risk factors in the child, would prompt the earlier institution of drug therapy after a trial of dietary intervention.
TREATMENT
Desired Outcomes
The goals of therapy expressed as LDL-C levels and the level of initiation of TLC and drug therapy are provided in Tables 11-8 and 11-9 for adults and children, respectively. While these goals are surrogate end points, the primary reason to institute TLC and drug therapy is to reduce the risk first or recurrent events such as MI, angina, heart failure, ischemic stroke, or other forms of peripheral arterial disease such as carotid stenosis or abdominal aortic aneurysm.
TABLE 11-9 Cut Points for Total Cholesterol and LDL Concentrations in Children and Adolescents
General Approach
Establishing targeted changes and outcomes with consistent reinforcement of goals and measures at followup visits to attain goals is important to reduce barriers for optimizing TLC and pharmacologic therapy.1 ![]() TLC should be implemented in all patients prior to considering drug therapy. The components of TLC include reduced intakes of saturated fats and cholesterol, dietary options to reduce LDL such as plant stanols and sterols and increased soluble fiber intake, weight reduction, and increased physical activity. In general, physical activity of moderate intensity 30 minutes/day for most days of the week should be encouraged.42,43 Patients with known CAD or at high risk should be evaluated before undertaking vigorous exercise. Weight and BMI should be determined at each visit and lifestyle patterns to induce a weight loss of 10% should be discussed in persons who are overweight. All patients should also be counseled to stop smoking and to meet the Joint National Committee VII guidelines for control of hypertension.
TLC should be implemented in all patients prior to considering drug therapy. The components of TLC include reduced intakes of saturated fats and cholesterol, dietary options to reduce LDL such as plant stanols and sterols and increased soluble fiber intake, weight reduction, and increased physical activity. In general, physical activity of moderate intensity 30 minutes/day for most days of the week should be encouraged.42,43 Patients with known CAD or at high risk should be evaluated before undertaking vigorous exercise. Weight and BMI should be determined at each visit and lifestyle patterns to induce a weight loss of 10% should be discussed in persons who are overweight. All patients should also be counseled to stop smoking and to meet the Joint National Committee VII guidelines for control of hypertension.
Nonpharmacologic Therapy
Individualized diet counseling that provides acceptable substitutions for unhealthy foods and ongoing reinforcement by a registered dietitian are necessary for maximal effect. The objectives of dietary therapy are to progressively decrease the intake of total fat, saturated fatty acids (i.e., saturated fat), and cholesterol, and to achieve a desirable body weight. Typical American diets now include 13% to 20% of total calories from saturated fat and a cholesterol intake of 350 to 450 mg/day, both in excess of a “heart-healthy” diet for normal Americans, let alone patients with a lipid disorder. Excessive dietary intake of cholesterol and saturated fatty acids lead to decreased hepatic clearance of LDL and deposition of LDL and oxidized LDL in peripheral tissues. The targeted saturated fatty acids have carbon chain lengths of 12 (lauric acid), 14 (myristic acid), and 16 (palmitic acid). The rationale for using a nutritionally balanced low-fat, low-cholesterol diet for the treatment of hypercholesterolemia is based on the following principles: (a) it represents a reasonable extension of the diet recommended for the general public; (b) it progressively decreases the major cholesterol-raising constituent of the diet; (c) it precludes large intakes of polyunsaturated fats; and (d) it facilitates weight reduction by removing foods of high caloric density.44–47
Dietary expertise in providing a wide range of options and suggestions in preparation of food can make the difference between a good and an inadequate response to diet. Information concerning eating out in a healthy fashion and advice for shopping are also important factors for success in diet therapy. An example is being aware of products with misleading labels such as coffee creamers that state they contain “no cholesterol,” when they may contain hydrogenated (saturated) fats or oils (e.g., palmitic acid, palm kernel oil, or coconut oil), which makes them undesirable because of their saturated fat content. Variations in polyunsaturated and saturated fat and cholesterol intake influence the LDL concentration, but the amount of cholesterol has been found to have a greater effect than the proportion of polyunsaturated or saturated fat. There were also racial differences in elevation of LDL with high saturated fat diets being greater in whites than in other racial groups. The isomeric form of fatty acids is also important.44 Fatty acids with the cis configuration are the preferred substrate for the ACAT reaction and significantly increase hepatic LDL-R clearance while reducing LDL cholesterol production rate. The trans isomeric form cannot be used by ACAT and is biologically inactive with no effect on LDL concentration.
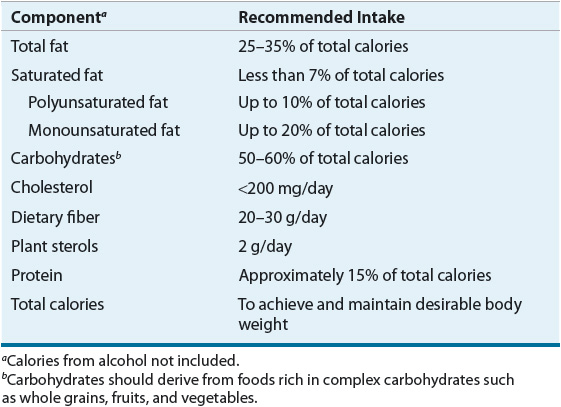
Ideally, therapeutic TLC including reduced intake of saturated fats and cholesterol, increased stanol/sterol and fiber intake, weight reduction, and increased physical activity should be used to attain lower LDL-C and to achieve reductions in CHD risk (Table 11-10). TLC may obviate the need for drug therapy, augment LDL-lowering drug therapy, and allow for lower doses. Weight control plus increased physical activity reduces risk beyond LDL cholesterol lowering, is the primary management approach for the metabolic syndrome, and raises HDL and reduces non-HDL cholesterol.48,49 Many persons should be given a 3-month trial (two visits spaced 6 weeks apart) of dietary therapy and TLC before advancing to drug therapy unless patients are at very high risk (severe hypercholesterolemia, known CHD, CHD risk equivalents, multiple risk factors, strong family history). Although changes in blood lipid levels may change before 3 months, adoption of a different eating pattern may require a longer period of time. It is important to involve all family members, especially if the patient is not the primary person preparing food. Both the NCEP and AHA have excellent Internet-based resources to aid patients in altering their diet in a culturally sensitive manner (http://www.americanheart.org/presenter.jhtml?identifier=1200009; http://www.nhlbi.nih.gov/health/index.htm). If all of the recommended dietary changes from NCEP, the estimated reduction, on average, in LDL would range from 20% to 30%.1 Adherence to diet and interindividual variability in macronutrient intake would obviously influence the eventual LDL level achieved. Based on the NHANES data, less than one half of the patients who should be instructed on heart-healthy diet receive any dietary instructions.
TABLE 11-10 Macronutrient Recommendations for the TLC Diet
Other dietary interventions or diet supplements may be useful in certain patients with lipid disorders. Increased intake of soluble fiber in the form of oat bran, pectins, certain gums, and psyllium products can result in useful adjunctive reductions in total and LDL cholesterol, but these dietary alterations or supplements should not be substituted for more active forms of treatment. Total daily fiber intake should be about 20 to 30 g/day, with about 25% or 6 g/day being soluble fiber.1 Studies with psyllium seed in doses of 10 to 15 g/day show reductions in total and LDL cholesterol ranging from about 5% to 20%.50,51 They have little or no effect on HDL-C or triglyceride concentrations. These products may also be useful in managing constipation associated with the bile acid sequestrants. Psyllium binds cholesterol in the gut and also reduces hepatic production and clearance. Fish oil supplementation provides an increased amount of the omega-3 polyunsaturated fatty acids such as eicosapentaenoic acid and docosahexaenoic acid. In epidemiologic studies, ingestion of large amounts of cold water, oily fish is associated with a reduction in CHD risk, but it is unclear whether the same advantage is conferred with commercially prepared fish oil products. Each 20 g/day ingestion of fish lowers CHD risk by 7% and eating fish once weekly or more should reduce CHD mortality.52 Fish oil supplementation has a fairly large effect in reducing triglycerides and VLDL-C, but it either has no effect on total and LDL cholesterol or may cause elevations in these fractions. Other actions of fish oil may account for their protective effects. These effects include quantitative and qualitative alterations in the synthesis of prostanoid substances, changes in immune function and cellular proliferation, and potential antioxidative actions.53 Responses noted with fish oil are further discussed in Pharmacologic Therapy below.54
Fat substitutes such as olestra (Olean, sucrose polyester, Procter and Gamble), a mixture of hexa-, hepta-, and octa-esters formed from the reaction of sucrose with long-chain fatty acids, are approved by the FDA as a nondigestible, nonabsorable, noncaloric fat substitute for snack foods. Olestra is heat stable, an advantage over several other fat substitutes, enabling it to be used in the preparation of fried and baked foods. It is similar in composition to triglycerides, but olestra is not hydrolyzed in the GI tract by pancreatic lipase, and, consequently, is not taken up by the intestinal mucosa. The principal adverse effects associated with olestra use are bloating, flatulence, diarrhea, and “anal leakage.” Because of the ability of olestra to solubilize lipophilic substances, there has been concern over potential drug interactions in which lipophilic drugs (e.g., cyclosporin, or colchicine) or vitamins (vitamins A, D, E, and K) are solubilized in olestra and excreted in the feces.
Recent studies have demonstrated the LDL-lowering effect of plant sterols, which are isolated from soybean and tall pine-tree oils. Ingestion of 2 to 3 g/day will reduce LDL by 6% to 15%.1 Plant sterols can be esterified to unsaturated fatty acids (creating sterol esters) to increase lipid solubility. Hydrogenating sterols produces plant stanols and, with esterification, stanol esters. The efficacy of plant sterols and plant stanols is considered to be comparable. Because lipids are needed to solubilize stanol/sterol esters, they are usually available in commercial margarines. The presence of plant stanols/sterols is listed on the food label. When margarine products are used, persons must be advised to adjust caloric intake to account for the calories contained in the products. Benecol® (McNeil), as an example, is a butter-like spread that contains a plant stanol ester, an ingredient that can lower cholesterol and that is derived from plant stanols found naturally in small amounts in foods such as wheat, rye, and corn.55 In August 2007, the FDA issued a warning about the consumption of red yeast rice and red yeast rice/policosanol-containing products. These products contained lovastatin that could interact with other drugs and would have the same toxicity of statins but would not be recognized by the consumer and the reduction in LDL is minimal.56
Drug therapy is indicated following an adequate trial of TLC changes as outlined in Tables 11-8 and 11-9.
Pharmacologic Therapy
There are now numerous randomized, double-blinded clinical trials demonstrating that reduction of LDL reduces CHD event rates in primary prevention, secondary intervention, and angiographic trials.57 Generally speaking, for every 1% reduction in LDL, there is a 1% reduction in CHD event rates.1 However, if treatment extends beyond the typical duration of a clinical trial (2 to 5 years), the accumulated benefit could be greater. Elevations of HDL of 1% result in approximately 2% reduction in CHD events.13,58 Of interest, angiographic trials, which typically cause small changes in luminal diameter (e.g., about a 0.04-mm difference in change between placebo and active treatment), result in fewer clinical events such as MI or the need for revascularization. This unexpected finding suggests that plaque size and luminal encroachment by plaque may be less important than the effects that cholesterol lowering may have on the activity in the plaque and endothelial dysfunction. These studies provide a strong rationale for attempting to lower plasma cholesterol and LDL in patients with hypercholesterolemia.
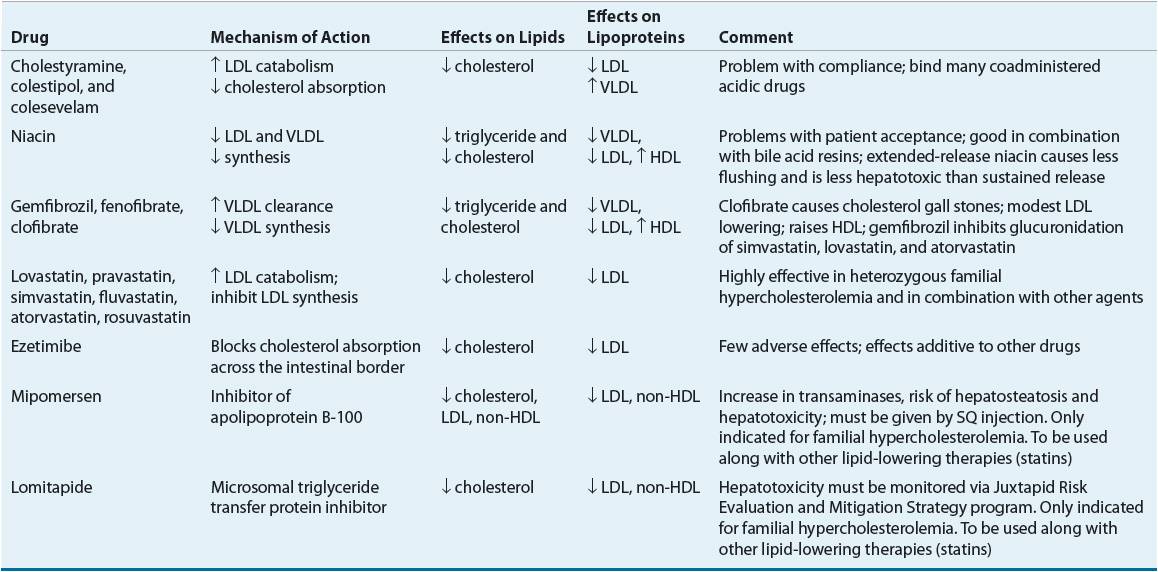
![]() Although many efficacious lipid-lowering drugs exist, none is effective in all lipoprotein disorders, and all such agents are associated with some adverse effects.59 Lipid-lowering drugs can be broadly divided into agents that decrease the synthesis of VLDL and LDL, agents that enhance VLDL clearance, agents that enhance LDL catabolism, agents that decrease cholesterol absorption, agents that elevate HDL, or some combination of these characteristics (Table 11-11). Table 11-12 lists recommended drugs of choice for each lipoprotein phenotype and alternate agents. Table 11-13 lists available products and their doses.
Although many efficacious lipid-lowering drugs exist, none is effective in all lipoprotein disorders, and all such agents are associated with some adverse effects.59 Lipid-lowering drugs can be broadly divided into agents that decrease the synthesis of VLDL and LDL, agents that enhance VLDL clearance, agents that enhance LDL catabolism, agents that decrease cholesterol absorption, agents that elevate HDL, or some combination of these characteristics (Table 11-11). Table 11-12 lists recommended drugs of choice for each lipoprotein phenotype and alternate agents. Table 11-13 lists available products and their doses.
TABLE 11-11 Effects of Drug Therapy on Lipids and Lipoproteins