Hypercatabolic States1
Stephen F. Lowry†
Susette M. Coyle
† Deceased.
1Abbreviations: ACTH, adrenocorticotropic hormone; AIDS, acquired immunodeficiency syndrome; CNS, central nervous system; GH, growth hormone; HIV, human immunodeficiency virus; IFN-γ, interferon-γ; IL, interleukin; IL-1ra, interleukin-1 receptor antagonist; REE, resting energy expenditure; T3, triiodothyronine; T4, thyroxine; TNF, tumor necrosis factor-α.
The hypercatabolic state is induced by endogenous cellular and systemic mediators produced in response to diverse stimuli, such as acute trauma and severe infection (sepsis), that are associated with increases in systemic metabolic rate and chronic inflammatory illnesses that promote only mild changes in metabolic rate. These conditions of metabolic stress are characterized by progressive, sometimes severe, loss of body protein and lipid stores, alterations in carbohydrate metabolism, and increased extracellular volume. Some hypercatabolic conditions may be ameliorated to some extent by nutritional support, but almost all require other interventions that promote palliation or cure of the underlying disease process. Although the hypercatabolic state implies a disruption of normal metabolic homeostasis, it also generally serves the teleologic purpose of healing and resolution of inflammation.
Since the pioneering work of Cuthbertson and Stewart (1) in 1945, the mechanisms underlying the host metabolic response to injury or catabolic diseases, as well as the means of modifying such responses via direct nutritional intervention or modulation of the inflammatory milieu, have been the subject of considerable investigation. This chapter describes the various phenotypes observed during these conditions, the neuroimmunoendocrine and cytokine mediators of this response, and the metabolic consequences of prolonged net hypermetabolism on the host. The clinical features of this response, in general, are qualitatively similar, regardless of the nature of the insult. However, the catabolic response to cancer and immunocompromised states, such as human immunodeficiency virus (HIV), are somewhat distinct and are reviewed separately.
METABOLIC RESPONSES TO INJURY AND CATABOLIC ILLNESSES
The temporal sequence of postinjury metabolic events is well known from the careful evaluation by many investigators. Cuthbertson and Stewart initially established the basis for understanding the biologic response to injury by determining that urinary excretion of nitrogen, potassium, and phosphorus was markedly increased in patients with long bone fractures. The relative concentrations of these excreted nutrients were similar to those of muscles, and he concluded that muscle was the source of such losses. These and subsequent isotopic dilution studies by Moore (2) characterized the ebb and flow phases of the postinjury response. The early ebb phase occurs immediately after insult and is characterized by hemodynamic instability with decreased cardiac output and oxygen consumption, low core temperature, and elevated glucagon, catecholamine, and free fatty acid levels. This phase typically lasts from 12 to 24 hours and is modified to some degree by the extent and composition of fluid resuscitation.
During the subsequent, more prolonged flow phase, increased total body oxygen consumption, metabolic rate, and amino acid efflux from peripheral muscle stores are evident, as are elevated counterregulatory hormone concentrations, urinary nitrogen losses, and varying degrees of peripheral tissue glucose intolerance (3) (Table 92.1). With effective resolution of the underlying inflammatory stimulus, a period of recuperation (anabolic phase) ensues during which metabolic homeostasis is reestablished in concert with replenishment of fat and muscle stores. Given that the greatest challenge to nutritional competency usually occurs during the flow phase during injury or illness, the mechanisms of this phase are discussed in more detail later.
TABLE 92.1 METABOLIC ALTERATIONS FOLLOWING INJURY | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Energy Expenditure
Injury and Sepsis
During the postinsult flow phase, total body oxygen consumption increases in concert with increased oxidation of fuel sources (carbohydrates, amino acids, and lipids). To some extent, the elevation of the metabolic rate correlates with the cause and/or the severity of the initial injury. Energy expenditure may increase minimally with mild injury, 15% to 25% after long bone fractures, and as much as double with burn injury over more than 40% of total body surface (3) (Fig. 92.1). The physiologic consequences of the flow phase insult serve as the basis for modern critical care medicine in which key features such as support of cardiac hemodynamics, optimized ventilation strategies, fluid administration, monitoring of organ function, and nutrition are aggressively supported in the seriously ill patient.
The increased metabolic rate necessitates mobilization of the endogenous nutrient stores to provide substrates for increased energy demand. The stores of carbohydrates, primarily glycogen, are depleted within the first 24 hours after injury. Thereafter, in the absence of exogenous nutrient sources, fat and protein provide the main energy sources. An obligatory net protein loss also exists in the hypermetabolic state that partly serves to provide substrates for gluconeogenesis and amino acids for increased synthesis of acute phase proteins. During the postinjury flow phase, an increase in urinary nitrogen excretion occurs that is generally roughly correlated to the magnitude of the injury and the adequacy of insult resolution. The nitrogen loss is primarily in the form of urea, but other contributing losses are in the form of creatinine, ammonia, uric acid, and amino acids excreted in urine. Skeletal muscle represents the majority of protein-containing tissues, and the net protein loss characteristic of the catabolic response results in loss of lean muscle mass. Stores of triglycerides are also mobilized and oxidized to provide substrates for the hypermetabolic state but are unable to prevent protein catabolism.
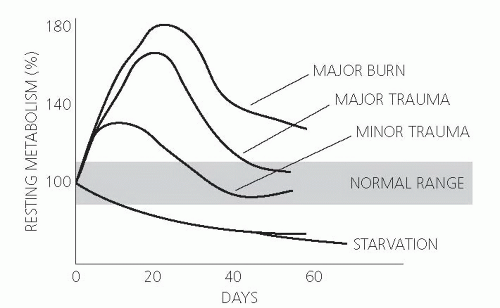 Fig. 92.1. Effect of injury on metabolic rate. (Adapted with permission from Wilmore DW. The Metabolic Management of the Critically Ill. New York: Plenum Medical Book, 1977.) |
A marked increase in the counterregulatory hormones occurs during the ebb and flow phases following acute injury: glucocorticoids, catecholamines, and glucagon mediated in large part by the central nervous system (CNS). The CNS responds to peripheral injury signals elicited by molecules derived from injured cells (4, 5) or, in the case of infection, the conserved molecular patterns of pathogens (6). The integrated systemic response to injury is effected by inflammatory mediators, such as cytokines, that influence cellular and systemic functions by both endocrine and paracrine mechanisms. These acute alterations in metabolic and hormonal responses serve to maintain tissue functions (Fig. 92.2).
Cancer
In contrast with the hypermetabolic state observed after trauma, hypermetabolism is not an invariable finding in patients with cancer. Because patients in the advanced stages of cancer often become cachectic, it was hypothesized that this weight loss results from a period of relative increase in energy expenditure and net negative energy balance. Some studies indicate a variable metabolic response to cancer; some investigators report a hypermetabolic response, whereas others observe no change or a hypometabolic response (7, 8, 9, 10) (Fig. 92.3).
Several studies show that the variations in resting energy expenditure (REE) seen in patients with cancer may be influenced by tumor histology (11, 12, 13). Elevated adrenergic activity and sensitivity has been demonstrated in tumor-bearing animals and patients with cancer (14) and may result, in part, from comorbidity factors such as systemic inflammation, a stress response to starvation, and cancer-related anemia.
Human Immunodeficiency Virus Infection
Another patient population in which a hypermetabolic response is often observed is those with HIV infection. Several studies suggest that the REE is elevated in patients with early HIV infection and increases further with subsequent acquired immunodeficiency syndrome (AIDS) (15, 16, 17). This REE increase may occur in an asymptomatic patient with normal CD4 lymphocyte counts (18) and
secondary infections may cause even further elevations in the REE. A few patients with AIDS exhibit metabolic rates similar to that seen in starvation (17).
secondary infections may cause even further elevations in the REE. A few patients with AIDS exhibit metabolic rates similar to that seen in starvation (17).
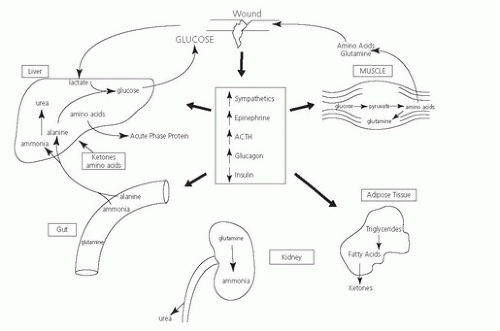 Fig. 92.2. Neuroendocrine and metabolic consequences of injury. ACTH, adrenocorticotropic hormone. |
Protein Metabolism
Injury and Sepsis
The large reserve of protein in skeletal muscle are rapidly mobilized in response to the increased energy demands after injury or acute inflammatory illnesses, and the extent of urinary nitrogen loss correlates with this activity. The accelerated rate of protein catabolism generally parallels the increase in oxygen consumption and represents a constant fraction of total oxidation after injury (3).
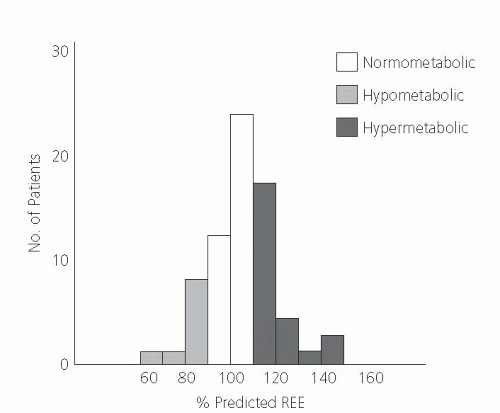 Fig. 92.3. Preoperative resting energy expenditure (REE) in patients with cancer. (Adapted with permission from Luketich JD, Mullen JL, Fuerer ID et al. Ablation of abnormal energy expenditure by curative tumor resection. Arch Surg 1990;125:337-41.) |
If allowed to proceed unabated, the net protein catabolism leads to loss of lean muscle mass and, in turn, may contribute to organ dysfunction or failure. Muscle myofibrillar protein is a prominent source of protein mobilization and reduced protein synthesis and inhibition of amino acid uptake by muscle is demonstrable (19). In animal models, important mediators of muscle catabolism include glucocorticoids, as well as certain cytokines, particularly tumor necrosis factor (TNF) and interleukin-1 (IL-1) (20). These and other inflammatory mediators that affect muscle protein metabolism can be produced by the myocyte or by nonmyocyte cells (i.e., macrophages), either locally at the site of injury or from a distance (21).
The cellular mechanisms involved in tissue protein breakdown in catabolic illness are not clearly defined. Animal studies indicate that under some catabolic conditions (e.g., sepsis), nonlysosomal, energy-dependent proteolysis may occur (22). This involves activation of the ubiquitin-proteasome-dependent pathway that increases mRNAs encoding ubiquitin and proteasomes in muscle. In this pathway, proteins destined for degradation are ligated to the polypeptide ubiquitin and then degraded by a protease that acts on ubiquinated proteins (23). The ubiquitination of proteins destined to undergo proteolysis by the proteasome is regulated by ubiquitin ligases as well as other enzymes. Ubiquitin ligases also appear to be involved in increased breakdown of proteins in atrophying muscle (24). Other studies indicated that
the mRNA for ubiquitin increases during glucocorticoid excess and acidosis, and this may occur in septic states (23). Additional proteases such as caspase-3 may release constituent proteins of actomyosin before ubiquitin-proteasome system activation (25). The ubiquitin pathway probably is important in proteolysis in many catabolic conditions, but further studies are needed to delineate the mechanisms leading to ubiquitin-proteasome system activation.
the mRNA for ubiquitin increases during glucocorticoid excess and acidosis, and this may occur in septic states (23). Additional proteases such as caspase-3 may release constituent proteins of actomyosin before ubiquitin-proteasome system activation (25). The ubiquitin pathway probably is important in proteolysis in many catabolic conditions, but further studies are needed to delineate the mechanisms leading to ubiquitin-proteasome system activation.
The increased amino acid efflux from peripheral stores after injury provides a substrate for enhanced hepatic gluconeogenesis and acute phase protein synthesis. It also allows synthesis of new protein tissue in wounds and proliferation of cellular components involved in the inflammatory response. Splanchnic uptake of glucogenic amino acids is increased, particularly alanine and glutamine. The increase in hepatic amino acid uptake in patients with burns corresponds with the quantity and distribution of peripheral tissue release (26). Enhanced splanchnic uptake is achieved by increased glutamine uptake in the gut enterocytes, whereas alanine is released by the gastrointestinal tract in increasing amounts during stress (27).
During the postinjury response, alanine and glutamine are also preferentially released from muscle stores. Although these amino acids make up approximately 6% of protein in muscle stores, they constitute 60% to 80% of the free amino acids released in response to insult (28, 29). Glutamine availability may become limited during catabolic illness, and mortality in critically ill patients has been associated with low glutamine levels (30).
Cancer
Patients with cancer and significant weight loss exhibit protein kinetics qualitatively similar to that observed in traumatized or infected patients. Whole body protein turnover rates are increased in some patients, with increases in protein synthesis as well as catabolism (31, 32). Attempts have been made to correlate the increase in protein turnover rates with changes in REE or weight loss. Isotope infusion studies suggest a significant increase in whole body protein catabolism in patients with cancer and cachexia compared with noncachectic patients with cancer or patients with benign disease (33). Although the whole body protein turnover rate is increased in other studies, it does not appear to correlate with energy expenditure or weight loss in most patients with cancer (8).
Human Immunodeficiency Virus Infection
In contrast with other patient populations, those infected with HIV in general show an elevated REE with decreased protein synthesis and catabolism in the absence of secondary infection. Preliminary data indicate that protein catabolism and a negative nitrogen balance occur when a patient with AIDS becomes secondarily infected (15). Despite effective antiretroviral therapy, the loss of lean body mass continues to be a common finding in patients with HIV infection, particularly in low-resource countries, and predicts morbidity and mortality (34, 35). Multiple factors are implicated in the loss of lean body mass, including reduced nutritional intake, nutrient malabsorption, elevated REE, and altered lipid, carbohydrate, and protein metabolism (17, 36, 37). Asymptomatic HIV-infected patients often do not exhibit a significant loss of lean body mass as compared with HIV-infected adults with secondary infections (38). This finding is in contrast with HIV-infected children, in whom growth failure secondary to low rates of lean tissue deposition may occur (39). These children have reduced protein balance, in part resulting from an inability to down-regulate protein catabolism.
Glucose Metabolism
Injury and Sepsis
Hyperglycemia is a common response to septic or traumatic injury. It results from both increased hepatic gluconeogenesis and decreased glucose uptake by insulindependent tissues. In the ebb phase, insulin levels are depressed, but they become normal to elevated during the flow phase, although remaining depressed in relation to the degree of hyperglycemia. The persistent hyperglycemia suggests injury-induced insulin resistance (3). In addition, studies using hepatic vein cannulations in thermally injured patients show increased uptake of gluconeogenic amino acids by the splanchnic tissues (26).
Stress-induced alterations in glucose metabolism result in decreased skeletal muscle uptake of glucose and decreased glucose incorporation into fatty acids by adipocytes (3). The decreased uptake by skeletal muscle is the result of peripheral insulin resistance, which may be mediated in part by excess cortisol and catecholamines (40, 41). In these stressed patients, hyperglycemia fails to suppress hepatic gluconeogenesis or glycogenolysis; and infusion of dextrose suppresses gluconeogenesis less effectively in patients with sepsis or trauma than in healthy volunteers (42). Amino acid infusions are also unable to inhibit gluconeogenesis in trauma patients (43).
During the stress response, another source of glucose results from the change to anaerobic glycolysis in skeletal muscle and hypoxic tissue (i.e., the wound) producing increased amounts of lactate. Lactate can be converted into glucose in the liver via the Cori cycle, which is increased in both burn and trauma patients (44). In patients with burns, lactate is a prominent gluconeogenic substrate. In critically ill patients, elevated lactate levels may reflect impaired tissue oxygenation; however, elevated lactate levels may persist despite evidence of adequate tissue oxygenation. The higher lactate levels in this circumstance reflect excess production of pyruvate as a consequence of accelerated glycolysis resulting from increased glucose uptake and glycogen breakdown rather than tissue hypoxia (45).
The efficiency of glucose oxidation is altered by trauma (46), surgical stress (45), and burn injury (47). Maximum glucose oxidative capacity appears to be inversely related to the severity of the injury. The decrease in oxidation may be the result of reduced activity of intracellular enzymatic metabolic pathways, such as pyruvate dehydrogenase (48).
Cancer
Glucose intolerance is often observed in association with malignant disease, and glucose intolerance has been documented in diverse cancer populations with advanced disease (49, 50, 51). This glucose intolerance may be related, in part, to other influences of cancer, such as weight loss, because glucose intolerance also occurs in response to weight loss attributable to calorie deprivation in benign disease (31).
Numerous reports note increased endogenous glucose production in patients with cancer (49, 52, 53), with an observed increase in glucose turnover rate affected by tumor stage (52, 54) and histology (50, 52). Studies comparing patients with early or advanced gastrointestinal malignant diseases indicated that rates of glucose turnover were significantly higher in patients with advanced lesions (50, 52, 54). Tumor histology may also influence glucose turnover rates. Patients with sarcoma (55) and leukemia (50) exhibit glucose turnover rates two to three times normal, whereas patients with lymphoma have a glucose turnover rate similar to that of normal subjects (50). Other studies indicate that the increased glucose turnover is related specifically to the cancer. The increase in glucose turnover rate is significantly higher in patients with cancer and weight loss than in the cancer-free population with comparable weight loss (31). Another study demonstrated that patients with cancer and progressive weight loss had markedly elevated rates of glucose turnover, whereas weight-stable patients with cancer had glucose turnover rates similar to those in normal volunteers (56). These findings contrast with the decreased glucose turnover rates in patients with weight loss attributable to uncomplicated undernutrition (54).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


