Fig. 13.1.
Down syndrome – brushfield spots on irides (A). Single transverse palmar creases and fifth finger clinodactyly (B). Gap between first and second toe (C).
♦
Developmental delay, learning disabled (IQ = 40–80), poor Moro reflex, infantile hypotonia, joint hyperflexibility, and increased risk for Alzheimer disease
♦
Congenital heart defects (endocardial cushion defects including atrioventricular canal defect and ventricular septal defects), increased risk for childhood acute leukemia, hypothyroidism, obesity, short stature, increased incidence of pulmonary hypoplasia, and duodenal atresia
♦
Ultrasound findings may include increased nuchal translucency, thickened nuchal fold, congenital heart defects, duodenal atresia, short humerus and femur, short middle phalanx of fifth finger, and echogenic bowel
♦
Seventy-five percent spontaneously abort in first trimester. Of those live born, 80% survival at age 30
Laboratory
♦
Chromosome analysis identifies extra chromosome 21
♦
Prenatal diagnosis through chromosome analysis of chorionic villi or amniocentesis
♦
An increased risk could be indicated by altered prenatal maternal serum markers or non-invasive prenatal screening (NIPS) using cell-free fetal nucleic acids
Treatment
♦
Not curable, supportive/symptomatic
Trisomy 13 (Patau Syndrome)
Chromosome and Gene Location
♦
Additional chromosome 13
Inheritance
♦
80% meiotic nondisjuncti on (47 chromosomes present)
80–90% maternal, increases with maternal age
10–20% paternal
♦
20% unbalanced translocation (46 chromosomes with an extra chromosome 13 fused to another acrocentric chromosome)
Incidence
♦
1/10,000 births
Clinical Manifestations
♦
Midline abnormalities ranging from simple ocular hypotelorism to cyclopia to complete absence of eyes, prominent occiput, microcephaly, malformed low-set ears, cl eft lip and palate, polydactyly, and transverse palmar crease (Fig. 13.2)
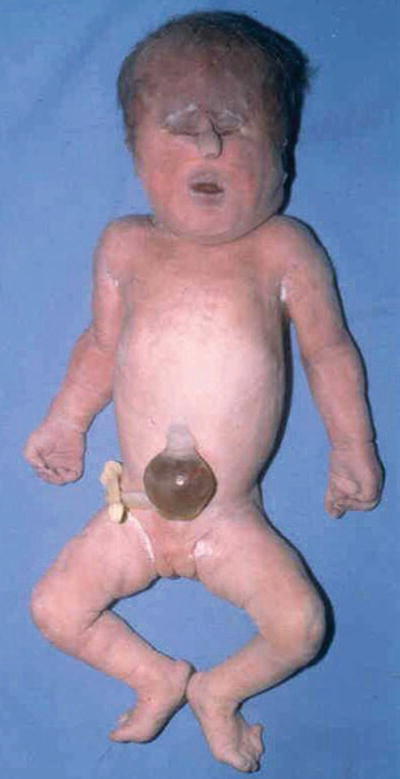
Fig. 13.2.
Trisomy 13 – showing hypertelorism and tubelike nasal structure. Polydactyly on foot.
♦
Complete or incomplete holoprosencephaly, severe intellectual disability, seizures, deafness, hypotonia, and apneic spells
♦
Congenital heart defects (hypoplastic left heart and ventricular septal defects), urogenital defects, cryptorchidism, bicornuate uterus and hypoplastic ovaries, polycystic kidneys, umbilical hernia, and omphalocele
♦
Ultrasound findings may include holoprosencephaly, cleft lip and palate, cystic hygroma, polydactyly, congenital heart defects, cystic kidneys, and omphalocele
♦
95% spontaneously abort. Of those live born, 90% die within first year of life
Laboratory
♦
Chromosome a nalysis identifies extra chromosome 13
♦
Prenatal diagnosis through chromosome analysis of chorionic villi or amniocentesis
♦
An increased risk could be indicated by altered prenatal maternal serum markers or non-invasive p renatal screening (NIPS) using cell-free fetal nucleic acids
Treatment
♦
Not curable, supportive/symptomatic
Trisomy 18 (Edward Syndrome)
Chromosome and Gene Location
♦
Additional chromosome 18
Inheritance
♦
Meiotic nondisjunction
95% maternal, increases with maternal age
5% paternal
Incidence
♦
1/5,000–1/10,000 births
Clinical Manifestations
♦
Microcephaly with prominent occiput, micrognathia, malformed ears, clenched hands, second and fifth digits overlapping third and fourth (Fig. 13.3), rocker bottom feet, single transverse palmar crease, and hypoplastic nails
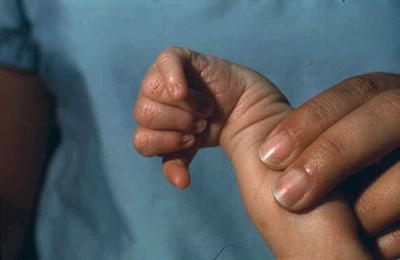
Fig. 13.3.
Trisomy 18 – clenched hand and overlapping fingers.
♦
Severe intellect ual disability, seizures, and hypertonia
♦
Severe intrauterine growth retardation, congenital heart defects (ventricular septal defects), urogenital defects, cryptorchidism, horseshoe kidney, diaphragmatic hernia, and omphalocele
♦
Ultrasound findings may include clenched hands, club and rocker bottom feet, micrognathia, congenital heart defects, omphalocele, diaphragmatic hernia, neural tube defects, ch oroid plexus cysts, and cystic hygroma
Life Expectancy
♦
95% spontaneously abort. Of those live born, 90% die within first year of life
Laboratory
♦
Chromosome analysis identifies extra chromosome 18
♦
Prenatal diagnosis through chromosome analysis of chorionic villi or amniocentesis
♦
An increased risk could be indicated by altered prenatal maternal serum markers or non-invasive pr enatal screening (NIPS) using cell-free fetal nucleic acids
Treatment
♦
Not curable, supportive/symptomatic
Klinefelter Syndrome (XXY)
Chromosome and Gene Location
♦
Extra X chromosome in a male
Inheritance
♦
Meiotic nondisjunction
55% maternal nondisjunction
45% paternal nondisju nction
♦
Also may be mosaic XY/XXY or rarely XX/XXY
Incidence
♦
1/800 males
Clinical Manifestations
♦
Tall habitus, undervirilized, small testes, gynecomastia, and poor musculature
♦
Mild delay, behavioral immaturity, shyness, learning disabilities (reading) speech delay
♦
Infertility
♦
Normal life expectancy
Laboratory
♦
Chromosome analysis identifies XXY
♦
Prenatal diagnosis through chromosome analysis of chorionic villi or amniocentesis
♦
Maternal serum screening and ultrasound findings are not useful
♦
An increased risk could be indicated by non-invasive prenatal screening (NIPS) using cell-free fetal nucleic acids
Treatment
♦
Testosterone suppleme ntation for the development of secondary sexual characteristics
Turner Syndrome (45, X)
Chromosome and Gene Location
♦
Missing or structurally abnormal X chromosome
Inheritance
♦
55% 45,X
80% loss of paternal X chromosome
20% loss of maternal X (no maternal age effect)
♦
25% 46,XX
Structural alt eration in one X chromosome
♦
15% mosaic
45,X with 46,XX, 46,XY, or others
Incidence
♦
1/2,000–1/5,000 female births
♦
Most common chr omosome finding in spontaneous abortions
Clinical Manifestations
♦
Short stature, webbed neck (Fig. 13.4), lymphedema of hands and feet, high arched palate, cystic hygroma, low posterior hairline, and hypoplastic widely spaced nipples
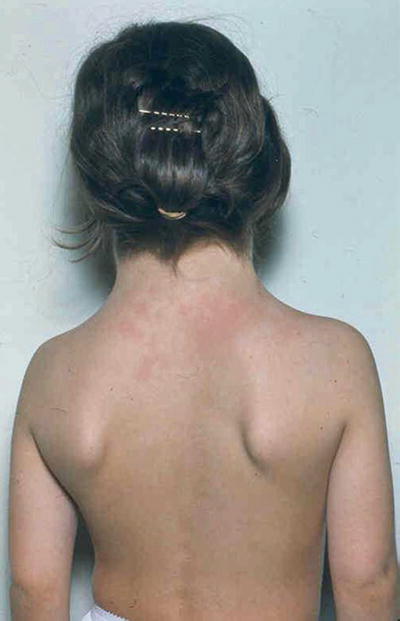
Fig. 13.4.
Turner syndrome – webbing of the neck and low posterior hairline.
♦
Normal or near normal intelligence; may have delay in speech, neuromotor skills, and learning abilities
♦
Gonadal dysgenesis (infertility, primary amenorrhea), renal malformations (horseshoe kidney), cardiovascular malformations (coarctation of aorta, hypoplastic left heart), and increased risk f or gonadoblastoma if mosaic for some Y chromatin
♦
Ultrasound findings include cystic hygroma (detectable after 10 weeks), lymph collections (ascites, pleural effusion), congenital heart disease, and renal anomalies
♦
99% spontaneously abort; those who survive infancy usually reach adulthood
Laboratory
♦
Chromosome analysis indicates monosomy X or other variants
♦
Prenatal diagnosis thro ugh chromosome analysis of chorionic villi or amniocentesis
♦
An increased risk could be indicated by altered prenatal maternal serum markers or non-invasive prenatal screening (NIPS) using cell-free fetal nucleic acids
Treatment
♦
Estrogen, thyroid, and growth hormone replacement therapy for development of secondary sexual characteristics and growth
Microdeletion Syndromes
♦
See Table 13.1
Table 13.1.
Microdeletion Syndromes
Syndrome | Features | Location |
|---|---|---|
Angelman | Ataxia, seizures, happy demeanor, severe intellectual disability | 15q11–q13 |
Chromosome 1p36 deletion | Intellectual disability, dysmorphic, hypotonia, seizures | 1p36 |
Chromosome 22q11.2 deletion (DiGeorge, velocardiofacial) | Cleft palate, heart defect, developmental delay, thymic and parathyroid hypoplasia | 22q11.2 |
Cri du chat | “Cat-like” cry as newborn, microcephaly, intellectual disability | 5p15.2 |
Miller–Dieker | Lissencephaly, intellectual disability | 17p13.3 |
Prader–Willi | Hyperphagia, obesity, intellectual disability, hypogonadism, small hands and feet | 15q11–q13 |
Smith–Magenis | Sleep disturbances, self-injurious behaviors, intellectual disability | 17p11.2 |
Williams | Supravalvular aortic stenosis, intellectual disability, hypercalcemia, social personality | 7q11.2 |
Wolf–Hirschhorn | High, broad nasal bridge, microcephaly, growth deficiency, clefting, hypertelorism, intellectual disability | 4p16 |
Angelman Syndrome
Chromosome and Gene Location
♦
15q11.2
Inheritance
♦
Angelman syndrome results from the loss of the maternally imprinted region at chromosome 15q11.2. Loss can occur via numerous mechanisms. Recurrence is dependent on mechanism of loss
60–70% deletion of maternal 15q11.2
5% paternal uniparental disomy (UPD) (two copies of the paternal chromosome)
3% imprinting defect
11% ubiquitin-protein ligase E3A (UBE3A) mutation
11% unknown
Incidence
♦
1/20,000
Clinical Manifestations
♦
Prominent mandibl e, open-mouthed expression, hyperreflexia, microcephaly, brachycephaly, and optic atrophy
♦
Ataxia, severe intellectual disability, seizures, and gross motor developmental delay
♦
Absent speech, inappropriate laughter, arm flapping, and feeding difficulties
♦
Most survive into adulthood
Laboratory
♦
First-tier testing
Molecular methylation anal ysis identifies missing maternal allele
Methylation-sensitive multiple ligation-dependent probe amplification (MLPA) detects missing maternal allele or deletion
♦
Second-tier testing
Fluorescent in situ hybridization (FISH) or chromosomal microarray (CMA) identifies deletion
Molecular UPD studies identify two copies of paternal allele
Sequence analysis identifies m utations in UBE3A gene
Mutation analysis of imprinting center
Treatment
♦
Not curable, supportive/symptomatic
Prader–Willi Syndrome
Chromosome and Gene Location
♦
15q11.2
Inheritance
♦
Prader–Willi syndrome results from the loss of the paternally imprinted region at chromosome 15q11.2. Loss can occur via numerous mechanisms. Recurrence is dependent on mechanism of loss
75% deletion of paternal 15q11.2
20–25% maternal UPD (two copies of maternal chromosome 15)
Remainder is thought to be due to imprinting defects
Incidence
♦
1/10,000–1/25,000
Clinical Manifestations
♦
Hypotonia, hypogon adism, obesity, small hands and feet, almond-shaped eyes, and short stature
♦
Mild to moderate intellectual disability
♦
Failure to thrive and hyperphagia
♦
Most survive into adulthood
Laboratory
♦
First-tier testing
Molecular methylation analysis id entifies missing paternal allele
Methylation-sensitive MLPA detects missing paternal allele or deletion
♦
Second-tier testing
FISH and/or CMA identifies deletion
Molecular UPD studies ident ify two copies of maternal allele if either first-tier test is abnormal
Mutation analysis of imprinting center
Treatment
♦
Not cu rable, supportive/symptomatic
1p36 Microdeletion Syndrome
Chromosome and Gene Location
♦
1p36
Inheritance
♦
90–95% sporadic
♦
5–10% familial translocations
Incidence
♦
1/5,000–10,000
Clinical Manifestations
♦
Microcephaly, deep-set eyes, flat nasal bridge, cardiac malformations, and hypotonia
♦
Intellectual disa bility and seizures
♦
Growth retardation
♦
Most survive into adulthood
Laboratory
♦
Deletion may or may not be detected on chromosome analysis depending upon resolution of the study and size of the deletion
♦
FISH and/or CMA identifies deletion
Treatment
♦
Not curable, supportive/sympto matic
Cri Du Chat Syndrome
Chromosome and Gene Location
♦
5p15
Inheritance
♦
85–90% sporadic (85% deletion, 4% mosaics, 3% ring chromosomes, 4% translocations)
♦
10–15% familial trans locations, inversions, and parental mosaicism
Incidence
♦
1/50,000
Clinical Manifestations
♦
“Cat-like” cry, microcephaly, hypertelorism, micrognathia, transverse palmar crease, and hypotonia
♦
Intellectual di sability
♦
50% no speech, growth failure
♦
Most survive into adulthood
Laboratory
♦
Deletion may or may not be detected on chromosome analysis depending upon resolution of the study an d size of the deletion
♦
FISH and/or CMA identifies deletion
Treatment
♦
Not curable, supportive/sympto matic
22q Deletion Syndrome (DiGeorge/Velocardiofacial Syndrome)
Chromosome and Gene Location
♦
22q11.2
Inheritance
♦
90% are deletions involving multiple genes
♦
Approximately 10–15% are familial (autosomal dominant)
Incidence
♦
1/4,000
Clinical Manifestations
♦
The disturbance of neural crest migration of pharyngeal pouches is thought to cause clinical features
♦
Interpatien t variability may be dependent on extent of deletion
♦
Hypertelorism, down-slanting eyes, high arched palate, micrognathia, low-set ears, bulbous nose, square nasal tip, cleft palate (velocardiofacial (VCF)), small open mouth, retrognathia, microcephaly, and slender hands and digits
♦
Cardiac manifestations include tetralogy of Fallot, outflow tract defect, right-sided aortic arch, interrupted aortic arch, and ventricular septal defect
♦
Mild-moderate learning dif ficulties, seizures, tetany, and emotional and behavioral problems
♦
Hypoparathyroidism, neonatal hypocalcemia (DiGeorge (DGS)), immune/T-cell deficit (DGS), hypernasal speech, hypospadius, and short stature
♦
Most reach ad ulthood if cardiac lesion is not life threatening
Laboratory
♦
Hypocalcemia, decreased T cells
♦
Deletion of 22q11 is usually not visible on chromosome analysis
♦
FISH and/or CMA identifies deletion
Treatment
♦
Cardiac surg ery, calcium supplements, and supportive care
Smith–Magenis Syndrome
Chromosome and Gene Location
♦
17p11.2
Inheritance
♦
Most are sporadic interstitial deletions
♦
A few cases of pericentric inversions with breakpoints in 17p11
Incidence
♦
1/50,000
Clinical Manifestations
♦
Brachycephaly, flat, broad midface, and prominent forehead
♦
Seizures and intellectual disability
♦
Hyperactivity, sleep disturbances, behavioral problems including screaming outbursts and self- mutilation behaviors, and speech delay
♦
Most survive into adulthood
Laboratory
♦
Chromosome analysis detects deletion in most cases
♦
FISH and/or CMA identifies deletion
Treatment
♦
Not curable, supportive/sympto matic
Miller–Dieker Syndrome
Chromosome and Gene Location
♦
17p13.3
Inheritance
♦
90% are sporadic deletions
♦
Approximately 10–15% are familial
Incidence
♦
1/100,000
Clinical Manifestations
♦
Lissencephaly, microcephaly, anteverted nostrils, carp mouth, and agenesis of corpus callosum
♦
Seizures and intellectual disability
♦
Failure to thrive and absent speech
♦
Life expectancy is variable, but most die in childhood
Laboratory
♦
Deletion may or may not be detected on chromosome analysis depending upon resolution of the st udy and size of the deletion
♦
FISH and/or CMA identifies deletion
Treatment
♦
Not curable, supportive/sympt omatic
Williams Syndrome
Chromosome and Gene Location
♦
7q11.23
Inheritance
♦
Mostly sporadic
♦
Few cases of autoso mal dominant inheritance have been reported
Incidence
♦
1/20,000 live births
Clinical Manifestations
♦
Broad forehead, bitemporal narrowness, periorbital fullness, wide mouth, broad nasal tip, long philtrum, micrognathia, growth retardation, and small widely spaced teeth
♦
Cardiac manifestations include supravalvular aortic stenosis and peripheral pulmonary stenosis
♦
Intellectual disability
♦
Gregarious personality, joint limitations, and hypercalcemia
♦
Most survive in to adulthood
Laboratory
♦
Elevated serum calcium
♦
Deletion is usua lly not visible on chromosome analysis
♦
FISH and/or CMA identifies deletion
Treatment
♦
Not curable, supportive/symptomatic
♦
Eliminatio n of vitamin D and calcium from the diet
Wolf–Hirschhorn Syndrome
Chromosome and Gene Location
♦
4p16.3
Inheritance
♦
85–90% sporadic
♦
10–15% familial translocations
Incidence
♦
1/50,000
Clinical Manifestations
♦
Microcephal y, hypertelorism, micrognathia, and hypotonia
♦
Seizures and intellectual disability
♦
Congenital heart defects, renal malformations, and genital malformations
♦
Most survive into adulthood
Laboratory
♦
Deletion may or may not be detected on chromosome analysis depending upon resolution of the study and size of the deletion
♦
FISH and/or CMA identifies deletion
Treatment
♦
Not curable, supportive/symptoma tic
Chromosome Breakage Syndromes
Fanconi Anemia
Chromosome and Gene Location
♦
Genetically heterogeneous (multiple gene loci involved)
Inheritance
♦
Autosomal recessive
Incidence
♦
1/22,000
Clinical Manifestations
♦
Short stature, ab sent or hypoplastic radii and thumbs, brown skin pigmentation, cryptorchidism, and renal anomalies
♦
Pancytopenia, anemia, increased incidence of leukemia, and solid tumors
Molecular Basis of Disease
♦
Multiple complementation groups have been identified and appear to be involved in the formation of a protein complex that participates in a DNA damage response pathway. The exact molecular mechanism that leads to impaired genomic stability has not been elucidated
Laboratory
♦
Increased chromosomal breakage, gaps, and rearrangements after exposure to diepoxybutan e or mitomycin C (DNA alkylating agents)
♦
Mutation analysis is available for specific mutations
Treatment
♦
Transfusions and bone marrow transplantation
♦
Not cura ble, supportive/symptomatic
Bloom Syndrome
Chromosome and Gene Location
♦
15q26.1
Inheritance
♦
Autosomal recessive
Incidence
♦
Rare
Clinical Manifestations
♦
Short s tature
♦
Facial erythema
♦
Increased susceptibility to infections
♦
Increase incidence of leukemia
♦
High pitched voice
Molecular Basis of Disease
♦
Decreased activity of DNA ligase I leads to genomic instability and multisystem anomalies
Laboratory
♦
Increased sister chromatid exchange (12× normal)
♦
Quadrilateral formation is increased as are random breaks and translocations between nonhomologous chromosomes
Treatment
♦
Not curable , supportive/symptomatic
♦
Minimize exposure to radiation/mutagenic agents
Ataxia Telangiectasia
Chromosome and Gene Location
♦
11q22–q23, ATM gene
Inheritance
♦
Autosomal recessive
Incidence
♦
1/40,000–1/100,000
Clinical Manifestations
♦
Cerebellar a taxia, conjunctival telangiectasia, and IgA deficiency
♦
Predisposition to malignancy
♦
Increased infections
♦
Growth failure, onset first 2 years of life
Molecular Basis of Disease
♦
Defect in DNA repair mechanisms leading to chromosomal breakage, increased intrachromosomal recombination, sensitivity to ionizing radiation (IR), and abnormal resistance to inhibition of DNA synthesis by IR
Laboratory
♦
Increased chromosomal breakage and X-radiation sensitivity; especially with breakpoints at sites of immunoglobulin genes or receptors
♦
7;14 chromosomal translocation is identified in 5–15% of cells
♦
Molecular analysis of ATM gene identifies mutation in approximately 95% of patients
Treatment
♦
Not curable, supportive/symptomatic
♦
Treatment of i nfections and neoplasms and avoidance of radiation
Xeroderma Pigmentosum
Chromosome and Gene Location
♦
Multiple loci
Inheritance
♦
Autosomal recessive
Incidence
♦
1/250,000
Clinical Manifestations
♦
Sensitivity to sunlight (blistering and freckling with little exposure, beginning in childhood)
♦
Predisposition to malignancy (especially skin cancer)
♦
Mental deterioration in some
Molecular Basis of Disease
♦
Defect in ult raviolet-induced DNA repair mechanisms
♦
Skin cells unable to repair sunlight-induced DNA damage
Laboratory
♦
Diagnosis is based on clinical criteria; no routine clinical laboratory abnormality is observed in patients with XP
♦
Cytogenetic analysis can identify clones of cells with chromosome abnormalities, increased ultraviolet-induced chromosome breaks, and sister chromatin exchanges
♦
Fibroblasts show UV hyp ersensitivity and abnormal unscheduled DNA synthesis
Treatment
♦
Not curable, supportive/symptomatic
♦
Avoidance of u ltraviolet light
Trinucleotide Repeat Disorders
♦
A growing number of inherited disease syndromes (primarily neurologic) are known to be caused by the abnormal presence of an expanded tract of trinucleotide repeats within disease-specific genes (Table 13.2)
Table 13.2.
Trinucleotide Repeat Disorders
Disease | Gene | Gene location | Repeat sequence | Repeat location, type of region | Normal allele size (no. of repeats) | Expanded mutant allele (fully penetrant) |
|---|---|---|---|---|---|---|
Fragile X syndrome | FMR1 | Xq | CGG | Noncoding region of Exon 1, 5′ untranslated region | 5–44 | >200 |
Myotonic dystrophy type 1 | DMPK | 19q | CTG | 3′ untranslated region | 5–34 | 50 to >2,000 |
Myotonic dystrophy type 2 | CNBP/ ZNF9 | 3q | CCTG | Intron 1 | 11–26 | 75–11,000 |
Friedreich ataxia | FXN | 9q | GAA | Intron 1 | 5–33 | 66–1,700 |
Huntington disease | HTT | 4p | CAG | Exon 1, polyglutamine coding | 10–26 | >40 |
Spinocerebellar ataxia type 1 | ATXN1 | 6p | CAG | Exon 8, polyglutamine coding | 19–38 | >38 |
Spinocerebellar ataxia type 2 | ATXN2 | 12q | CAG | Exon 1, polyglutamine coding | 14–31 | >32 |
Spinocerebellar ataxia type 3 | ATXN3 | 14q | CAG | Exon 10, polyglutamine coding | <44 | 52–86 |
Spinocerebellar ataxia type 6 | CACNA1A | 19p | CAG | 3′ End of gene, polyglutamine coding | 4–18 | 20–33 |
Spinocerebellar ataxia type 7 | ATXN7 | 3p | CAG | Exon 1, polyglutamine coding | 4–19 | >36 |
Spinal and bulbar muscular atrophy | AR | Xq | CAG | Exon 1, polyglutamine coding | 11–34 | >37 |
♦
Typically, the clinical hallmark of these trinucleotide repeat diseases is anticipation. Anticipation is defined as the clinical observance of an earlier age of onset and increased rate of disease progression due to amplification in the number of expanded repeats in successive generations
♦
The diagnosis is made by determining the number of trinucleotide repeats within a specific disease-causing expandable allele (within the proper clinical context). The normal allele will have a “normal range” of trinucleotide repeats, whereas the disease-associated (expanded) allele will contain an increased number of repeats (in the hundreds and thousands, for some diseases)
Fragile X Syndrome
Chromosome and Gene Location
♦
Xq27.3
Inheritance
♦
X-linked d ominant
Incidence
♦
1/5,000 males (accounts for up to 5% of male intellectual disability)
♦
1/2,500 females
Clinical Manifestations
♦
Intellectual disability
♦
Large narrow face, prominent forehead and jaw, with moderately increased head circumference, and large ears
♦
Macroorc hidism (80%)
Molecular Basis of Disease
♦
Expanded CGG repetitive element within FMR1 gene
5–44 normal
45–54 intermediate (minimal expansion due to meiotic instability in a small percentage of cases), no associated phenotype
55–200 premutation (meiotic instability with potential to expand to full mutation), fragile X-associated tremor/ataxia syndrome (FXTAS) characterized by cerebellar ataxia with the observation of white matter lesions on MRI and intentional tremor is observed in both males and females with almost half of all male premutation carriers affected by age 79; other clinical manifes tations of FXTAS include memory loss, cognitive deficit, parkinsonism, and neuropathy; premature ovarian failure (POF) is also observed in an average 21% of female premutation carriers; however, penetrance is inversely correlated with the number of repeats in the intermediate and premutation carrier ranges
>200 full mutation, leads to abnormal methylation and transcriptional suppression of FMR1 gene and absence of FMRP (RNA binding protein)
CGG repeat expansion is through female germ line (males transmit premutation repeat without much change)
♦
Sherman paradox
Describes apparent deviation from traditional Mendelian inheritance and varies according to whether the causative gene is transmitted through a male or female
In fragile X, this is the result of expansion from premutation to full mutation through the female germ line only
Laboratory
♦
Molecular analysis (PCR and Southern blot with methylation analysis) detects expanded CGG repeat and is the recommended first-tier molecular test, as expanded CGG repe ats account for ~99% of disease-causing mutations
♦
Molecular analysis by sequencing and dosage analysis detect the remaining 1% of FMR1 mutations
♦
Chromosomal fragile site may be apparent by standard chromosome analysis when culture d in folate-deficient/thymidine inhibiting media
Treatment
♦
Not curable
♦
Supportive/symptomatic
Myotonic Dystrophy Type 1
Chromosome and Gene Location
♦
19q13.2
Inheritance
♦
Autosomal dominant
Incidence
♦
1/8,000 (DM1 and DM2, together, represent the most common muscular dystrophy in adults)
Clinical Manifestations
♦
Myotonia (sustained muscle contraction), muscle wasting, and facial weakness
♦
Cataracts
♦
Hypogonadism
♦
Frontal balding
♦
Cardiac conduction disturbances
♦
Diabetes mellitus (5%)
♦
Swallowing and speech disability
♦
Respiratory failure
♦
Neonatal hypotonia
♦
Delayed motor development
♦
Wide phenotypic range and age of onset from severely affected infants (congenital DM1) to classically affected adults (classic DM1) to minimally symptomatic elderly individuals (mild DM1) with mildly affected adults having myotonia and cataracts vs congenital DM1 which is characterized by severe weakness and often fatal respirato ry insufficiency
Molecular Basis of Disease
♦
Expansion of a trinucleotide CTG repeat in t he myotonic dystrophy protein kinase (DMPK) gene
5–34 normal allele size
35–49 mutable normal (premutation) allele; not associated with disease
>50 full penetrance allele; severity of phenotype increases as repeat size increases (mild DM1: 50–150 repeats; classic DM1: 100–1,000 repeats)
>1,000 full penetrance allele, severely affected (congenital form)
Expansion occurs preferentially through mate rnal transmission but can also occur through paternal transmission
Laboratory
♦
Molecular analysis identifies expanded repeat in nearly 100% of cases
♦
Electromyogram demonstrates characteristic electrical myotonic discharges
♦
Muscle biopsy ma y show marked proliferation of fibers within the central nuclei, sacroplasmic masses, and type 1 muscle fiber atrophy
Treatment
♦
Not curable, supportive/symptomatic
♦
Monitor and treat for cataracts, cardiac conduction disturbances, diabetes, sleep apnea, hyp ogonadism, and other endocrine problems
Myotonic Dystrophy Type 2 (Proximal Myotonic Myopathy)
Chromosome and Gene Location
♦
3q21.3
Inheritance
♦
Autosomal dominant
Incidence
♦
1/8,000 (DM1 and DM2; together, th e most common muscular dystrophy in adults)
Clinical Manifestations
♦
Myotonia (sustained muscle contraction), proximal weakness primarily in the thighs, and muscle pain
♦
Cataracts
♦
Cardiac conduction disturbances
♦
Insulin insensitivity predisposing to hyperglycemia and diabetes mellitus
♦
Hypogonadism
♦
Onset typically occurs in the third decade of life
♦
Myotonic dystrophy type 2 includes DM2 and proximal myotonic myopathy (PROMM)
Molecular Basis of Disease
♦
Expansion of a trinucleotide CCTG repeat in intron 1 of the CNBP/ZNF9 gene
Reported allele sizes usually include the total number of repeats/base pairs in the (TG) n (TCTG) n (CCTG) n repeat tract all present at the DM2 locus
Tetranucleotides (TCTG or GCTG) commonly interrupt the CCTG repeats in the normal range; however, these sequence interruptions are not found in fully expanded pathogenic alleles
11–26 CCTG repeats (104–176 bp total allele length) normal allele
27–74 CCTG repeats (177–372 bp total allele length) intermediate normal (intermediate/borderline alleles of unclear significance) allele; never been reported
75–11,000 CCTG repeats (372–44,000 bp total allele length) full penetrance allele
CCGT repeat size typically co ntracts through either maternal or paternal transmissions and somatic instability causes CCGT repeat sizes to increase as the affected individual ages
Phenotype and age of onset cannot be predicted by CCGT repeat size as there is no correlation between repeat size and se verity of disease
Laboratory
♦
Molecular analysis identifies expanded repeat in 99% of cases
♦
Electromyogram demonstrates characteristic electrical myotonic changes
♦
Muscle biopsy may show marked proliferation of fibers within the central nuclei and ty pe 2 muscle fiber atrophy
Treatment
♦
Not curable, supportive/symptomatic
♦
Monitor a nd treat for cataracts, cardiac conduction disturbances, diabetes, and hypogonadism
Friedreich Ataxia
Chromosome and Gene Location
♦
9q21.11
Inheritance
♦
Autosomal recessive
Incidence
♦
1/25,000–1/50,000
Clinical Manifestations
♦
Progressive ataxia, muscle weakness, dysarthria, and dysphagia
♦
Absent deep tendon reflexes and decreased vibration and/or joint-position sense
♦
Optic nerve atrophy (25%)
♦
Deafness (13%)
♦
Scoliosis
♦
Hypertrophic cardiomyopathy (66%)
♦
Bladder dysfunctio n (urinary frequency and urgency)
♦
Hammer toes, pes cavus (foot deformity marked by high arch), and restless leg syndrome
♦
Diabetes (30%) or glucose intolerance (50%)
♦
Obstructive sleep apnea (20%)
♦
Age of onset is approximately 10–15 years for classic Friedreich ataxia (FRDA); however, ~15% of cases are associated with delayed onset (late-onset FRDA, LOFA, and very late-onset FRDA, VLOFA), and ~12% are associated with delayed-on set and intact tendon reflexes (FRDA with retained reflexes, FARR)
Molecular Basis of Disease
♦
Expansion of a trinucleotide GAA repeat in intron 1 of the frataxin (FXN) gene
5–33 normal allele
34–65 premutation (meiotic instability with potential to expand to full penetrance allele) allele. (Note: the term borderline alleles, defined as 44–66, is sometimes used because the shortest repeat le ngth associated with disease has not yet been clearly determined)
66–1,700 full penetrance allele
Carriers of permutation alleles are very rare; therefore, expansion of alleles from one generation to the next is typically not observed, and anticipation is not a hallmark characteristic of this disease
Statistically significant correlations have been observed between the size of the smaller of the two expanded alleles and age of onset (i.e., LOFA and VLOFA), presence of leg muscle weakness, duration of wheelchair use, and presence of other clinical manifestations
Laboratory
♦
Molecular analysis identifies two expanded repeat sizes in 90–94% of cases
♦
Molecular analysis by sequencing and dosage analysis detects the remaining FXN mutations
♦
Nerve conduction studies generally show slow or absent sensory nerve conduction velociti es but normal motor nerve conduction
Treatment
♦
Not curable, supportive/symptomatic
♦
Monitor for hypertrophic cardiomyopathy (by echocardiography and electrocardiogram), diabetes, and deafness
♦
Treat with assistive devices for weakness, antispasmodic agents for bladder dysfunction, and speech therapy to maximize communication
Huntington Disease
Chromosome and Gene Location
♦
4p16.3
Inheritance
♦
Autosomal dominant
Incidence
♦
3/100,000–7/100,000 (Western European ancestry)
♦
0.1/100,000–15/100,000 (range, all ethnicities)
Clinical Manifestations
♦
Slowly progress ive fatal disorder with onset of neurologic or psychiatric changes usually in mid-life (35–44 years)
♦
Life expectancy is approximately 50–60 years or about 15–18 years after initial onset of disease
♦
Early (preclinical)
Behavioral/mood changes and personality changes (72%)
Depression
Anxiety
Delusions and hallucinations
Abnormal eye movements
Minor changes in coor dination and movement
♦
Middle (clinical)
Chorea (90%)
Involuntary movements
Difficulty with voluntary movements (slow, difficult to initiate or control, impaired reaction time, trouble with balance and walking)
Weakness
Dysarthria and dysphagia
Cognitive decline
♦
Late
Inability to walk and speak
Incontinence
Rigidity and dystonia
Total dependence on others
♦
Juvenile Huntington disease (onset prior to age 20 years) is characterized by motor, cognitive, and psychiatric changes; however, the clinical presentation differs and is much more severe than in adults; epileptic seizures are also common
Molecular Basis of Disease
♦
Disease results from an expansion of a trinucleotide CAG repeat in exon 1 of the HTT gene
10–26 normal allele
27–35 intermediate allele (meiotic instability, expansion to full penetrance allele may occur with transmission); not associated with disease
36–39 reduced penetrance allele (at risk but not certain to develop symptoms of disease)
>40 full penetrance allele
>60 full penetrance allele is associated with juvenile onset Huntington disease
Expansion can occur through both maternal and paternal transmission; however, large expansions are more frequentl y observed through paternal transmission
Inverse correlation between number of CAG repeats and age of onset
Laboratory
♦
Molecula r analysis identifies expanded repeat in 100% of cases
♦
Imaging studies such as magnetic resonance imaging (MRI) identifies atrophy of the caudate nucleus and putamen
Treatment
♦
Not curable, supportive/symptom atic
Autosomal Dominant Cerebellar Ataxias
Chromosome and Gene Location
♦
Numerous loci have been identified, and more than 30 subtypes have been delineated (commonly referred to as spinocerebellar ataxias, SCAs are numbered by subtype with the exception of DRPLA; dentatorubropallidoluysian atrophy)
Inheritance
♦
Autosomal d ominant
Incidence
♦
1–5/100,000
Clinical Manifestations
♦
Clinical manifestations vary by subtype; however, the most common features include adult-onset gait ataxia, dysarthria, ataxia (NOS), visual disturbance and diplopia, and dizziness
♦
Peripheral neuropathy
♦
Death usually occurs 10–20 years following age of onset
Molecular Basis of Disease
♦
Many subtypes of spinocerebellar ataxia (SCA) result from expansion of trinucleotide CAG repeat, a lthough some (e.g., SCA5) result from nonrepeat mutations
♦
SCA3 (also known as Machado–Joseph disease) is the most common subtype followed by SCA 1, 2, 6, and 7 (represented in Table 13.2)
Laboratory
♦
Molecular an alysis identifies expanded trinucleotide repeat in most subtypes
♦
Neuroimaging may be necessary to distinguish hereditary forms of ataxia from acquired forms
Treatment
♦
Not curable, supportive/symptoma tic
Spinal and Bulbar Muscular Atrophy (Kennedy Disease)
Chromosome and Gene Location
♦
Xq12
Inheritance
♦
X-linked recessive
Incidence
♦
1/50,000 males
Clinical Manifestations
♦
Teen to adult onset, usually in the third to fifth decades of life
♦
Slowly progressive proximal muscle weakness, muscle atrophy, and fasciculations with bulbar involvement, dysarthria, and dysphagia
♦
Androgen insensitivity (gynecomastia, reduced fertility, testicular atrophy)
♦
Normal life expectancy
Molecular Basis of Disease
♦
Expansion of trinucleotide CAG repeat in t he human androgen receptor (AR) gene
11–34 normal allele size
35 unknown clinical significance
36–37 reduced penetrance allele (at risk but not certain to develop symptoms of disease)
>37 full penetrance allele
The larger the expansion, the earlier the age at onset of disease and the more rapid the disease progression
Laboratory
♦
Molecula r analysis identifies expanded repeat in 100% of cases
Treatment
♦
Not curable, supportive/symptomatic
♦
Hormone replacement as needed
♦
Monitor with annual strength testing and pulmonary function
Neuromuscular Disorders
Duchenne/Becker Muscular Dystrophy
Chromosome and Gene Location
♦
Xp21.2
Inheritance
♦
X-linked r ecessive
Incidence
♦
1/3,500 (male) 1/1,500 (female carriers)
30% of cases are new mutations
5–15% of sporadic cases are result of gonadal mosaicism (mother carries a subpopulation of oocytes with the mutation)
Clinical Manifestations
♦
Duchenne muscular dystrophy
Proximal muscle weakness le ading to difficulties involving gait, jumping, and climbing stairs
Positive for Gower sign (use of arms to push oneself into standing position by moving hands up one’s thighs, indicative of hip weakness)
Pseudohypertrophy of the calf muscles and proximal muscle weakness
Rapidly progressive with loss of ability to walk before age 13
Dilated cardiomyopathy (100% after by age 18)
Intellectual disability (25–35%)
Death usually occurs by the third decade in most
♦
Becker muscular dystrophy
Milder course of muscle involve ment with later-onset skeletal muscle weakness
Slower progression, wheelchair dependent after age 16
Survival into the 30–40 s, with dilated cardiomyopathy being the most common cause of death
Cognitive problems ar e rare
♦
Female carriers of Duchenne/Becker muscular dystrophy (DMD or BMD)
Muscle weakness (19% DMD carriers, 14% BMD carriers)
Myalgia cramps (5%)
Left ventricle dilation (19% DMD carriers, 16% BMD carriers)
Dilated cardiom yopathy (8% of DMD carriers)
Molecular Basis of Disease
♦
Dystrophin is a protein found in the sarcolemma of normal muscle. It is thought to be involved in the anchorin g of the cytoskeleton of the muscle cell to extracellular proteins
♦
DMD and BMD result from alterations within the dystrophin (DMD) gene
♦
Deletions in 50–70% of DMD and BMD
Deletions that disrupt the reading frame of th e triplet code (frameshift mutations) lead to DMD
Deletions that do not disrupt the reading frame of the triplet code (in-frame mutations) most often lead to BMD
♦
Duplications in 5–10% of DMD and BMD
♦
Point mutati ons in 20–35% of DMD and 10–20% of BMD
Laboratory
♦
Pathology
Variability in size of muscle fiber s, degeneration, atrophy of individual fibers, and proliferation of endomysial and perimysial connective tissue
♦
Antidystrophin antibodies detect
<5% of normal dystrophin in DMD
5–20% in mild DMD or severe BMD
>20% normal dystrophin co rrelates with BMD
♦
Serum creatinine phosphokinase (CK) concentration
>5–10× the normal range (100% of affected individuals)
50% of DMD carriers and 33% of BMD carriers have elevated CK levels (2–10× normal)
Caution must be used as CK levels vary with age, pregnancy, and activity
♦
Genetics
Deletions and duplications detected directly by molecular analysis
Linkage analysis is available when deletion/duplication analysis negative
Treatment
♦
Not curable, su pportive/symptomatic
♦
Treatment of dilated cardiomyopathy with medication and transplantation in rare cases
♦
Corticoste roid therapy (prednisone) in affected individuals to improve strength and function
Spinal Muscular Atrophy, Types I–IV
Chromosome and Gene Location
♦
5q13.1
Inheritance
♦
Aut osomal recessive
Incidence
♦
1/25,000
Clinical Manifestations
♦
See Table 13.3
Table 13.3.
Spinal Muscular Atrophy (SMA) Subtypes
Type I (Werdnig–Hoffmann) | Type II (Dubowitz disease) | Type III (Kugelberg–Welander) | Type IV |
|---|---|---|---|
Onset 0–6 months | Onset after 6 months | Onset after 10 months | Onset second or third decade of life |
Reduced fetal movements | Mild/arrested type 1 | Ambulation feasible | Muscle weakness |
General muscle weakness | Low muscle tone | Waddling gait | Independent ambulation |
Low muscle tone | Non-ambulatory | Muscle weakness | |
Respiratory muscle weakness | Finger trembling | Fasciculations | |
Arthrogryposis | Absent tendon reflexes | Contractures | |
Tongue fasciculations | Increased life span when respiratory function preserved | ||
Contractures | |||
Lack of motor development | |||
Absence of tendon reflexes | |||
Death often by 1 year |
Molecular Basis of Disease
♦
Survival motor neuron (SMN1) gene is homozygously deleted in nearly all of types I and II and ab out 80% of type III spinal muscular atrophy (SMA)
♦
2–5% of type I SMA is caused by compound heterozygosity for an SMN1 deletion and point mutation
♦
The presence of three or more copies of SMN2, a gene just adjacent to SMN1, is associated with a milder SMA phenotype in individuals with two SMN1 deletions and/or point mutations
Laboratory
♦
The primary test used to diagnose individuals with SMA is molecular analysis for deletions in exon 7 and/or exon 8 of SMN1 gene
♦
Muscle biopsy reveals group atrophy of type 1 and type 2 muscle fibers
Treatment
♦
Not curable, supportive/symptomat ic
Charcot–Marie–Tooth Disease
Chromosome and Gene Location
♦
Numerous loci have been identified
Inheritance
♦
Autosomal dominant, recessive and X-linked forms (see Table 13.4)
Table 13.4.
Charcot–Marie–Tooth ( CMT) Subtypes
Disorder (% of CMT) | CMT1 (50 %) | CMT2 (20–40 %) | CMT4 (rare) | X-linked CMT (10–20 %) | |
|---|---|---|---|---|---|
CMT1A (70–80 %) | CMT1B (5–10 %) | ||||
Gene location | 17p | 1q | Multiple loci | Multiple loci | X |
Gene name | PMP22 | MPZ | Multiple genes | Multiple genes | GJB1 (90 %), PRPS1, other unknown genes |
Inheritance | Autosomal dominant | Autosomal dominant | Autosomal dominant | Autosomal recessive | X-linked dominant |
Molecular genetics | 1.5 kb duplication of PMP22, point mutations in PMP22 | Point mutations in MPZ | Point mutation in NFL and HSPB1 | Point mutations in GDAP1, EGR2, or PDX | Point mutations in GJB1 |
Incidence
♦
1/3,300 (the mos t common genetic cause of neuropathy)
Clinical Manifestations
♦
Hereditary neuropathy resulting in progressive distal muscular atrophy and weakness of arms and legs (presenting in first to third decade)
♦
Pes cavus
♦
CMT1 – Charcot–Marie–Tooth type 1 (50%)
Demyelinating per ipheral neuropathy
Distal muscle weakness and atrophy
Decreased nerve conduction velocities
Absent deep tendon reflexes
Onset 5–25 years
Six subtypes (CMT1A–CMT1F) are dis tinguishable only by molecular analysis
Autosomal dominant inheritance
♦
CMT2 – Charcot–Marie–Tooth type 2 (20–40%)
Axonal (non-demyelinat ing) neuropathy
Distal muscle weakness and atrophy
Normal or slightly decreased nerve conduction velocities
Deep tendon reflexes are preserved
Milder phenotype than CMT1
15 subtypes distinguishable by molecular analysis
Autosomal dominan t inheritance
♦
Autosomal dominant intermediate CMT
Demyelinating and axonal neuropathy
Decreased nerve conduction velocities
♦
CMT4 – Charcot–Marie–Tooth type 4
Demyelinating or a xonal neuropathy
Distal muscle weakness and atrophy
Pes cavus
Autosomal recessive inheritance
♦
CMTX – X-linked Charcot–Marie–Tooth (10–15%)
Axonal neuropathy in males, intellec tual disability, deafness, and spasticity
Five subtypes
Molecular Basis of Disease
♦
Multiple gene s at different loci are involved (see Table 13.4). Molecular analysis is available for many subtypes. Some subtypes are clinically indistinguishable and are designated solely on molecular findings
Laboratory
♦
Molecular testing is available for many of the CMT subtypes
♦
Electrophysiological studies and nerve biopsy may be helpful in distinguishing it from other acquired and hereditary forms of peripheral neuropathy and in establishing a diag nosis of CMT
Treatment
♦
Not curable, supportive/symptomatic
Hereditary Neuropathy with Liability to Pressure Palsies
Chromosome and Gene Location
♦
17p11.2
Inheritance
♦
Autosoma l dominant
Incidence
♦
Unknown
Clinical Manifestations
♦
Recurrent transient palsies (i.e., carpel tunnel syndrome and foot drop)
♦
Sensory dysfunction (result of compression to peripheral nerve)
♦
Pes cavus (20%)
♦
Absent ank le reflexes (50–80%) and reduced tendon reflexes (15–30%)
♦
Scoliosis
Molecular Basis of Disease
♦
Deletion at 17p11.2 involving the peripheral myelin protein 22 (PMP22) gene (80%) or poin t mutations in the PMP22 gene (20%)
♦
Unequal crossing over between misaligned repetitive elements leads to the hereditary neuropathy with liability to pressure palsies (HNPP) deletion and the CMT1A duplication syndromes
♦
De novo mutations are most often paternally derived
Laboratory
♦
Cytogene tics using FISH or molecular genetics analysis identifies deletion at 17p11.2 involving the PMP22 gene or a point mutation
♦
Nerve biopsy reveals sausage-shaped swellings of myelin sheath
♦
Increase in distal motor latency of the median nerve at the wrist in both symptomatic and asymptomatic individuals
♦
Reduced motor and sensory nerve conduction velocity
Treatment
♦
Not curable, supportive/sympt omatic
Skeletal Disorders
Craniosynostosis
Apert Syndrome
Chromosome and Gene Location
♦
10q24 (FGFR2)
Inheritance
♦
Autosomal dominant, vast majority due to new mutation
Incidence
♦
1/65,000–1/100,000
Clinical Manifestations
♦
Brac hycephaly due to coronal craniosynostosis
♦
Hypoplasia of midface and hypertelorism
♦
“Mitten hand” deformity; symmetric and severe osseous or cutaneous syndactyly affecting hands and feet, and broad thumbs and great toes
♦
Fused ce rvical vertebrae, typically C5–C6
♦
Genitourinary anomalies in 10%
♦
Cardiac anomalies in 10%
♦
Intellectual disability
Molecular Basis of Disease
♦
Mutation in fibroblast growth factor receptor 2 (FGFR2), tyrosine kinase receptor involved in regulation of osteogenesis
♦
Two common gain-of-function mutations account for >98% of cases
Laboratory
♦
Targeted m olecular testing for common mutations, whole-gene sequencing, and deletion/duplication studies of FGFR2 are all available
Treatment
♦
Not curable, sup portive/symptomatic and surgical management
♦
Early surgical consultation and correction are important to reduce the risk of intracranial pressure
Crouzon Syndrome
Chromosome and Gene Location
♦
10q24 (FGFR2)
Inheritance
♦
Autosomal dominant with variable expressivity
Incidence
♦
1/25,000
Clinical Manifestations
♦
Coron al, lambdoid, or sagittal craniosynostosis
♦
Hypoplasia of midface
♦
Hypertelorism
♦
Proptosis
♦
Beaked nose
♦
Normal intelligence
Molecular Basis of Disease
♦
Mutation in FGFR2, tyrosine ki nase receptor involved in regulation of osteogenesis
Laboratory
♦
Targeted molecular testing for common mutations, sequencing of select exons or the entire FGFR2 gene, and deletion/duplication studies of FGFR2 are all available
Treatment
♦
Not curable, supp ortive/symptomatic and surgical management
Pfeiffer Syndrome (Types I–III)
Chromosome and Gene Location
♦
8p11.2 (FGFR1) type I
♦
10q24 (FGFR2) types I–III
Inheritance
♦
Autosom al dominant with variable expressivity
Incidence
♦
1/100,000
Clinical Manifestations
♦
See Table 13.5
Table 13.5.
Clinical Findings of Pfeiffer Syndrome
Craniofacial | Extremities | Intellect | Other | |
|---|---|---|---|---|
Type I | Brachycephaly (premature synostosis of coronal and often sagittal sutures) | Broad, medially deviated thumbs and great toes, variable syndactyly | Typically normal | Hearing loss, hydrocephalus occasionally |
Type II | Cloverleaf skull (premature synostosis of all but metopic and squamosal sutures), extreme proptosis | Broad, medially deviated thumbs and great toes, elbow ankylosis/synostosis | Developmental delay/intellectual disability commonly | Choanal stenosis/atresia, laryngotracheal anomalies, hydrocephalus, seizures |
Type III | Turribrachycephaly (premature synostosis of bicoronal, sagittal, and metopic sutures), extreme proptosis | Broad, medially deviated thumbs and great toes | Developmental delay/intellectual disability commonly | Choanal stenosis/atresia, laryngotracheal anomalies, hydrocephalus, seizures |
Molecular Basis of Disease
♦
Type I: mutation in fibroblast growth factor receptor (FGFR) 1 (5% of cases) or 2 (95% of cases), tyro sine kinase receptors involved in regulation of osteogenesis
♦
Type II or III: mutation in FGFR2, tyrosine kinase receptor involved in regulation of osteogenesis
Laboratory
♦
Targeted molecular testing for common mutations, sequencing of select exons or the entire FGFR1/FGFR2 genes, and deletion/duplication studies of FGFR1/ FG FR2 are all available
Treatment
♦
Not curable, supportive/symptomatic and surgical management
♦
Types II and III are typically more severe and have an increased risk for early death, but early and aggressive surgical and medical treatment may increase the possibility of a positive outcome
Saethre–Chotzen Syndrome
Chromosome and Gene Location
♦
7p21.1 (TWIST1)
Inheritance
♦
Autosomal dominant
Incidence
♦
1/25,000–1/50,000
Clinical Manifestations
♦
Brachyc ephaly due to coronal craniosynostosis
♦
Ptosis
♦
Maxillary hypoplasia
♦
Small ears with prominent crus
♦
Cutaneous syndactyly
♦
Normal intelligence typically
♦
Patients wi th microdeletions may have intellectual disability
Molecular Basis of Disease
♦
Typically caused by loss of function of the TWIST gene, which negatively regulates FGFR and osteogenic transcription factors. Loss-of-function mutations, exonic or whole-gene deletions, and translocations/inversions involving 7p21 have all been reported
Laboratory
♦
Targeted molecular testing for common mutations, sequencing of select exons or the entire gene, and deletion/duplication studies of TWIST1 are all available
Treatment
♦
Not curab le, supportive/symptomatic and surgical management
Muenke Syndrome
Chromosome and Gene Location
♦
4p16.3 (FGFR3)
Inheritance
♦
Autosomal dominant
Incidence
♦
1/30,000
Clinical Manifestations
♦
Brachyc ephaly due to bilateral coronal craniosynostosis or anterior plagiocephaly due to unilateral coronal craniosynostosis
♦
Intellectual disabilities
♦
Strabismus
♦
Hearing loss
♦
Carpal bone and/or tarsal bone fusions
♦
Broad toes and/or thumbs
♦
High-arche d palate or cleft lip and/or palate
Molecular Basis of Disease
♦
Specific pathogenic mutation p.Pro250Arg in FGFR3 which accelerates bone differentiation
Laboratory
♦
Targeted molecular testing for the common mutation, sequencing of select exons or the entire gene, and deletion/duplication studies of FGFR3 are all available
Treatment
♦
Not curable, supportive/sym tomatic
Achondroplasia
Chromosome and Gene Location
♦
4p16.3 (FGFR3)
Inheritance
♦
Autosoma l dominant; 80% result from a new mutation
Incidence
♦
1/25,000
Clinical Manifestations
♦
Short stature, proximal shortening of long bones, disproportionate shortening of limbs, and gen u varum
♦
Large head, frontal bossing, and hypoplasia of midface
♦
Infantile hypotonia
♦
Gross motor developmental delay
♦
Normal int elligence
♦
Normal life expectancy
♦
Also at risk for cord compression due to odontoid hypoplasia
Molecular Basis of Disease
♦
Mutation in transmembrane domain of fibroblast growth factor transmembrane receptor (FGFR3)
♦
>99% caused by same mutation
Laboratory
♦
X-ray implicates skeletal involvement
♦
Targeted mutation testing for the common mutations in FGFR3 that account for >99% of cases is available. Sequencing of select exons or the entire FGFR3 gene is also avai lable
Treatment
♦
Not curable, supportive/sympto matic
Osteogenesis Imperfecta (Types I–VII)
Chromosome and Gene Location
♦
See Table 13.6
Table 13.6.
Clinical Findings of Osteogenesis Imperfecta
Inheritance | Clinical findings | Gene | Abnormal collagen chains | |
|---|---|---|---|---|
Type I | Autosomal dominant | Bone fragility, blue sclera, hearing loss | COL1A1 | Pro-α1(I) |
Type II | Autosomal dominant, but usually new germ line mutation, 6–7% recurrence risk due to parental gonadal mosaicism | Perinatal lethal, calvarial under-mineralization, beaded ribs, compressed long bones, dark blue sclera | COL1A1 COL1A2 | Pro-a1(I) Pro-α2(I) |
Type III | Autosomal dominant Autosomal recessive (rarely) | Multiple prenatal bone fractures, limb shortening, limb deformities, deafness, blue sclera | COL1A1 COL1A2 | Pro-a1(I) Pro-α2(I) |
Type IV | Autosomal dominant | Mild short stature, mild deformity, dentinogenesis imperfecta, normal/gray sclera | COL1A1 COL1A2 | Pro-a1(I) Pro-α2(I) |
Type V | Autosomal dominant | Variable stature, multiple fractures, moderate bone deformities, normal sclera | Unknown | |
Type VI | Uncertain | Mild short stature, multiple fractures, rhizomelic shortening, normal sclera | Unknown | |
Type VII | Autosomal recessive | Mild short stature, multiple fractures, bone deformity, normal sclera | CRTAP |
Incidence
♦
1/20,000–30,000
Clinical Manifestations
♦
A genetic consulta tion is recommended given clinical and molecular genetic diagnosis of one of the many types may be quite complex
Molecular Basis of Disease
♦
Collagen is the major protein of the white fibers of connective tissue, cartilage, and bone
♦
There have been nu merous types of collagen identified
♦
Mutations in the procollagen genes, whose products make up the triple helix of type 1 collagen, lead to the various types of osteogenesis imperfecta
♦
Clinical presentation is dependent on the extent to which the mutation alters the protein product
Laboratory
♦
X-ray implicates s keletal involvement
♦
Molecular genetic testing of the procollagen and some of the other disease-causing genes is available
♦
Collagen testing on cultured fibroblasts available
Treatment
♦
Not curable, supportive/symptomatic
♦
Surgical inte rvention when indicated
Connective Tissue Disorders
Marfan Syndrome
Chromosome and Gene Location
♦
15q21.1 (FBN1)
Inheritance
♦
Autosomal dominant
♦
15–30% result from a new mutation
Incidence
♦
1 in 5,000–10,000
Clinical Manifestations
♦
Diagno sis based on clinical criteria
♦
Tall, thin habitus, long extremities, and arachnodactyly (Fig. 13.5A)
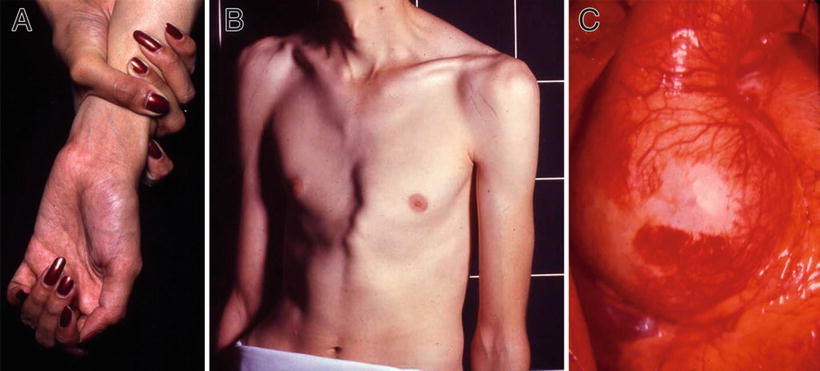
Fig. 13.5.
Marfan syndrome – arachnodactyly, wrist sign (A). Pectus deformity and striae on shoulders (B). Intraoperative view of dilated aortic root (C).
♦
Ectopia lentis and retinal detachment
♦
Without treatment, life expectancy is reduced to about two-thirds normal life span. With proper management of cardiovascular manifestations, life expectancy approximates that of the general population
Molecular Basis of Disease
♦
FBN1 codes for fibrillin, a structural protein, which is the major constituent of microfibrils
Laboratory
♦
Sanger sequencing a nd deletion/duplication testing via array comparative genomic hybridization (aCGH) or multiplex ligation-dependent probe amplification (MLPA) is available for the FBN1 gene
♦
FBN1 analysis is also available as a part of several clinically available multigene panels which utilize next-generation sequencing
Treatment
♦
Surgical inte rvention when indicated
♦
Close monitoring of heart defects as they can lead to sudden death
♦
Use of beta-adrenergic blockade
♦
Clinical trials underway to assess the potential benefits of angiotensin II receptor antagonist drugs (losartan) on slowing aneurysm progression
♦
Referral to genetics for personal and family history evaluation, detailed medical exam, and app ropriate health management
Loeys–Dietz Syndrome
Chromosome and Gene Location
♦
9q22 (TGFBR1), 3p22 (TGFBR2), 15q22.33 (SMAD3), and 1q41 (TGFB2)
Inheritance
♦
Autosomal dominant
♦
~75% result from new mutation
Incidence
♦
Unknown, no apparent enrichment in any ethnic group
Clinical Manifestations
♦
Minimal clinical diagnostic criteria not established
♦
Two clinical phenotypes described. LDS type I has vascular, skeletal, and craniofacial findings. LDS type II has vascular, skeletal, and cutaneous findings
♦
Dilatation or dissection of the aorta. Aortic dilatation presents in >95% of probands. Arterial aneurysms tend to be more aggressive and dissect at smaller aortic dimensions than in Marf an syndrome
♦
Other arterial aneurysms and tortuosity throughout the arterial tree
♦
Pectus excavatum or pectus carinatum and scoliosis
♦
Joint laxity
♦
Arachnodactyly
♦
Talipes equinovarus
♦
Ocular hyper telorism, bifid uvula/cleft palate, and craniosynostosis (most commonly sagittal, but also coronal and metopic) in LDS type I
♦
Translucent skin, easy bruising, and dystrophic scars in LDS type II
Molecular Basis of Disease
♦
Mutations in genes coding for a transforming growth factor ligand, cofactor, and two receptors, which regulate a variety of cellular processes, including proliferation, differentiation, cell cycle arrest, apoptosis, and formation of the extrac ellular matrix
Laboratory
♦
Sanger sequencing and deletion/duplication testing via array comparative genomic hybridization (aCGH) or multiplex ligation-dependent probe amplification (MLPA) is a vailable for TGFBR1, TGFBR2, SMAD3, and TGFB2
♦
TGFBR1, TGFBR2, SMAD3, and TGFB2 analysis is also available as a part of several clinically available multigene panels which utilize next-generation sequencing
Treatment
♦
Echocardiography at frequent intervals to monitor ascending aorta, MRA, or CT as indicated
♦
Early and aggressive surgical intervention
♦
Use of beta-adrenergic blockade
♦
Referral to genetics for personal and family history evaluation, detailed medical exam, and ap propriate health management
Thoracic Aortic Aneurysm and Dissection
Chromosome and Gene Location
♦
Seven genes and two loci have been associated with thoracic aortic aneurysm and dissection (TAAD):
TGFBR2 on 3p22
TGFBR1 on 9q33–q34
MYH11 on 16p13.13–p13, 12
ACTA2 on 10q22–q24
MYLK on 3q21
SMAD3 on 15q22.33
AAT2 (TAAD1) locus on 5q13–q14 (causative gene unknown)
AAT1 (FAA1) locus on 11q23.3–q24 (causative gene unknown)
Rare cases of FBN1 mutations, 15q21.1, have been associated with isolated TAAD (incidence unkn own)
Inheritance
♦
Autosomal dominant
Incidence
♦
Aortic aneurysms account for approximately 1–2% of deaths in industrialized society
♦
Approximately 20% of familial TAAD is accounted for by mutations in known genes. It is likely that other genes with variable penetrance have yet to be discovered
Clinical Manifestations
♦
Major diagnostic criteria for TAAD are the following:
♦
Presence of dilatation and/or dissection of the ascending thoracic aorta or dissection of the descending aorta just distal to the origin of the subclavian artery
♦
Exclusion of Marfan syndrome, Loeys–Dietz syndrome, and other connective tissue abnormalities
♦
Other occasional manifestations include inguinal hernia, scoliosis, aneurysms in other portions of the aorta, cerebral aneurysms, livedo reticularis (ACTA2), iris flocculi (ACTA2), bicuspid aor tic valve (ACTA2), and patent ductus arteriosus (MYH11)
Molecular Basis of Disease
♦
TGFBR1, TGFBR2, and SMAD3 code for proteins that regulate a variety of cellular processes, including proliferation, differentiation, cell cycle arrest, apoptosis, and formation of the extracellular matrix. Mutations in TAAD families are most often found in the kinase domain of these proteins, though rarely other mutations have been reported
♦
MYH11 codes for smooth muscle heavy chain protein. Smooth muscle myosin is the major contractile protein of smooth muscle and is composed of a MYH11 dimer and two pairs of nonidentical light chains
♦
ACTA2 codes for smooth muscle cell alpha-actin, a major contractile protein in smooth muscle cells
♦
MYLK codes for a phosphokinase which facilitates interactions between myosin and actin filaments, important in contractile activity
Laboratory
♦
Sanger sequencing and deletion/duplication testing via array comparative genomic hybridization (aCGH) or multiplex ligation-dependent probe amplification (MLPA) is available fo r TGFBR1, TGFBR2, MYH11, ACTA2, FBN1, and SMAD3
♦
Sanger sequencing is available for MYLK
♦
TGFBR1, TGFBR2, MYH11, ACTA2, MYLK, FBN1, and SMAD3 analysis is also available as a part of several clinically available multigene panels which utilize next-generation sequencing
Treatment
♦
Echocardio graphy to monitor aortic root, consider imaging entire aorta and other vasculature
♦
Prophylactic surgical repair of the aorta (timing depends on rate of progression, causative gene, etc)
♦
Medications that reduce hemodynamic stress, such as beta-adrenergic blockade
♦
Aggressive treatment of hypertension
♦
Assessmen t and standard treatment for hernias and scoliosis
♦
Referral to genetics for personal and family history evaluation, detailed medical exam, a nd appropriate health management
Ehlers–Danlos Syndrome
Chromosome and Gene Location
♦
Classic type: 9q34 (COL5A1), and 2q31 (COL5A2)
♦
Vascular type: 2q31 (COL3A1)
♦
Others
Hypermobility type: c ausative gene is mostly unknown; there are some cases of TNXB mutations (6p21)
Kyphoscoliotic type: 1p36 (PLOD1)
Arthrochalasia type: 17q21–q22 (COL1A1), and 7q22 (COL1A2)
Spondylocheiro dy splastic form: 11p11.2 (SLC39A13)
Dermatosparactic type: 5q35.3 (ADAMTS2)
Musculocontractural type 1: 15q15.1 (CHST14)
Progressive kyphoscoliosis, myopathy, and hearing loss type: 7p15 (FKBP14)
Inheritance
♦
Classic type, arthrochalasia type, and vascular type are autosomal dominant
♦
Most rare forms of Ehlers–Danlos syndrome are autosomal recessive
♦
Mutations in TNXB can be inherited in an autosomal dominant or autosomal recessive manner
Incidence
♦
Classi c type: estimated 1/20,000
♦
Vascular type: estimated 1/250,000
Clinical Manifestations
♦
Classic type
Skin hyperex tensibility
Smooth, velvety skin
Widened atrophic scarring
Abnormal, delayed wound healing
Joint hypermobility
Joint dislocations
Easy bruising
Generalized tissue fragility
Less common ly, mitral and tricuspid valve prolapse, aortic root dilatation, and spontaneous rupture of large arteries
♦
Vascular type
Thin, translucent skin
Characteristi c facial appearance (thin nose and lips, emaciated face with prominent cheekbones, eyes appear sunken or bulging, often with coloring around them and thin telangiectasia on the eyelids)
Easy bruising
Arterial, intestinal, and/or uterine fragility
Vascular rupture or dissection
GI perforation
Org an rupture
Molecular Basis of Disease
♦
Collagen is the major protein of the white fibers of connective tissue, cartilage, and bone
♦
Mutations in collagen genes lead to decreased synthesis, altered secretion, and instability of collagen
♦
The defect for some types is unknown
Laboratory
♦
Protein analysis is available for some subtypes
♦
Histological features are nondiagnostic
♦
Sanger sequencing and deletion/duplication testing via array comparative genomic hybridization (aCGH) or multiplex ligation-dependent probe amplification (MLPA) is available for COL5A1, COL5A2, COL3A1, COL1A1, and COL1A2
♦
Sanger sequencing is available for PLOD1
♦
ADAMTS2 , CHST14, COL1A1, COL1A2, COL3A1, COL5A1, COL5A2, FKBP14, PLOD1, and SLC39A13 analysis is also available as a part of several clinically available multigene panels which utilize next-generation sequencing
Treatment
♦
Not curable, supportive/symptomatic
♦
Referral to genetics for personal and family history evaluation, detailed medical exam, and a ppropriate health management
Stickler Syndrome
Chromosome and Gene Location
♦
12q13 ( COL2A 1) and 1p21 (COL11A1) account for the majority of individuals with Stickler syndrome
♦
Also associated with 6p21.3 (COL11A2), 6q13 (COL9A1), 1p33–p32 (COL9A2), and 20q13.3 (COL9A3)
Inheritance
♦
Autosomal dominant for COL2A1, COL11A1, and COL11A2
♦
Autosomal recessive for COL9A1, COL9A2, and COL9A3
Incidence
♦
While the exact prevalence is unknown, estimations based on incidence of Pierre Robin sequence in newborns and the percent of these newborns who develop signs or symptoms of Stickler syndrome are approximately 1 in 7,500–9,000
Clinical Manifestations
♦
Progressive myopia, retinal detachment, and blindness
♦
Pierre Robin sequence (micrognathia and abnormal smallness of the tongue, often with cleft palate)
♦
Severe myo pia, congenital glaucoma, retinal detachment, and cataracts
♦
Premature degenerative changes in various joints with abnormal epiphyseal development
♦
Mitral valve prolapse
Molecular Basis of Disease
♦
The COL2A1 gene encodes the chains of type II collagen, a major structural component of cartilag inous tissues
♦
The COL11A1 gene encodes the alpha 1 chain of type XI collagen, thought to play an important role in fibrillogenesis by controlling lateral growth of collagen II fibrils
♦
The COL11A2 gene encodes the alpha 2 chain of type XI collagen expressed in cartilage but not in adult liver, skin, tendon, or vitreous
♦
The COL9A1, COL9A2, and COL9A3 genes code for type IX collagen which, together, form a subunit of three alpha chains found in tissues containing type II collagen
Laboratory
♦
Skeletal X-ra ys show changes of a skeletal dysplasia
♦
Sanger sequencing and deletion/duplication testing via array comparative genomic hybridization (aCGH) or multiplex ligation-dependent probe amplification (MLPA) is available for COL2A1, COL11A1, COL11A2, COL9A1, COL9A2, and COL9A3
♦
COL2A1, COL11A1, COL11A2, COL9A1, COL9A2, and COL9A3 analysis is also available as a part of several clinically available multigene panels which utilize next-generation sequencing
Treatment
♦
Not curable, supportive/symptomatic
♦
Referral to geneti cs for personal and family history evaluation, detailed medical exam, and appropriate health management
Alport Syndrome
Chromosome and Gene Location
♦
Xq22.3 (COL4A5), 2q36–q37 (COL4A3), and 2q35–q37 (COL4A4)
Inheritance
♦
COL4A5 mutations are inherited in an X-linked pattern (80% total AS cases)
♦
COL4A3 and COL4A4 cases are inherited in both autosomal recessive (15% total AS cases) and autosomal dominant (5% total AS cases) patterns
Incidence
♦
Estimated to be 1 in 50,000 live births
Clinical Manifestations
♦
Renal failure
♦
Sensorineural deafness
♦
Lenticonus
♦
Macular changes
Molecular Basis of Disease
♦
Mutations in the typ e IV collagen genes result in abnormalities in expression of the collagen chains and absent or defective structure and function in the collagen networks of the basement membranes
♦
Molecular testing on clinical basis
Laboratory
♦
Microscopic hematuria
♦
Urinary red cell casts
♦
Proteinuria
♦
Leukocyturia
♦
Abnormal glomer ular basement membrane on electron microscopy
Treatment
♦
Referral to genetics for personal and family history evaluation, detailed medical exam, and appropriate health management
♦
Not curable, sup portive/symptomatic
♦
Kidney transplant as indicated
Hematologic Disorders
Alpha Thalassemia
♦
Normal adult hemoglobin (Hb A) is a tetramer of two alpha and two beta globin chains. The alpha thalassemias are a group of inherited conditions characterized by decreased synthesis of alpha globin chains, resulting in an imbalance of globin chains in the formation of the hemoglobin (Hb) tetramer
Chromosome and Gene Location
♦
16p13.3-pt er (there are two alpha globin genes present at this locus on both copies of chromosome 16 for total normal complement of four alpha globin genes)
Inheritance
♦
Complex: individuals with alpha thalassemia may have alterations in two, three, or four alpha globin genes
Incidence
♦
Varies by po pulation; most common in African-American, Southeast Asian, Mediterranean, and Indian populations
♦
Severe forms occur predominantly in Asians
Clinical Manifestations
♦
An individual with one altered alpha globin gene is a “silent” carrier and typically does not have any clinical symptoms (α+-thalassemia)
♦
Individuals with two altered alpha globin genes, either of different chromosomes (i.e., in trans) or on the same chromosome (i.e., in cis), have alpha thalassemia trait (α0-thalassemia), which manifests as minimal anemia with microcytosis
♦
Hemoglobin H (Hb H) disease results when three alpha globin genes are altered. Hb H is an abnormal tetramer of four beta chains and is unstable. Thus, Hb H disease is a form of hemolytic anemia. This is characterized by moderate anemia, enlarged liver and spleen, and erythroid hyperplasia in the bone marrow
♦
Bart hydro ps fetalis results when all four alpha globin genes are not functional. Hb Bart is a tetramer of four gamma chains. The oxygen affinity of hemoglobin Bart is so high that it cannot release oxygen to the tissues. Onset is in the fetal period and includes severe hypochromic anemia, extramedullary erythropoiesis, generalized edema, pleural and pericardial effusions, hepatosplenomegaly, and hydrocephalus. This condition is not compatible with life; death occurs from anoxia in utero
Molecular Basis of Disease
♦
Alpha thalassemia results from mutations in the HBA1 and HBA2 genes that encode α1-globin and α2-globin, respectively. Mutations result in reduced production of the alpha globin chains. The clinical severity is determined by the degree of alpha globin chain deficiency relative to beta globin production. Numerous mutations have been found
♦
The most common types of mutations are deletions of one or both alpha globin genes, which results from the misalignment and subsequent recombination of the alpha globin genes during meiosis (i.e., nonhomologous crossover)
Laboratory
♦
Red blood cell indices reveal microcytic anemia in Hb H and α-thalassemia trait. Silent carriers generally have normal red blood cell indices. Hb Bart disease shows macrocytic red blood cells
♦
Peripheral blood s mear demonstrates hypochromic red cells and anisopoikilocytosis in Hb Bart syndrome. In Hb H disease, microcytosis, hypochromia, anisocytosis, and poikilocytosis are apparent. Carriers have morphologic changes that are less severe than affected individuals and may also have reduced MCV and MCH
♦
Inclusion bodies can be shown in Hb H disease using supravital stain; carriers and those with alpha thalassemia trait can also show small amounts of inclusions
♦
Hemoglobin analysis (e.g., electrophoresis, HPLC, and isoelectric focusing) detects the presence of Hb H (the abnormal β-globin tetramer) in adults and Hb Bart (an abnormal ϒ-globin tetramer) in infants
♦
Molecular genetic analysis is clinically available
Treatment
♦
Hemolytic or aplastic crisis caused by Hb H disease can be treated with red blood cell transfusions
♦
Splenect omy may be performed in cases of massive splenomegaly
♦
Iron chelation therapy is used if chronic blood transfusions are necessary
♦
Surveillance includes hematologic evaluation and growth and development assessment. Iro n overload should be monitored in individuals requiring transfusions
Beta Thalassemia
Chromosome and Gene Location
♦
11p15.5
Inheritance
♦
Typically autosomal recessive
Incidence
♦
Most commo nly found in Mediterranean and Southeast Asian populations but also in Middle Easterners, Africans, and African Americans
Clinical Manifestations
♦
The clinical presentation and classification of beta thalassemia is dependent on the extent to which beta chain production is decreased and the resulting imbalance of alpha globin chains to non-alpha globin chains
♦
Beta thalasse mia becomes clinically evident as fetal hemoglobin production decreases after birth and adult hemoglobin fails to replace it (around 6 months of age)
♦
Beta thalassemia major results from the homozygous or compound heterozygous mutations that severely reduce beta chain production
Onset between 6 and 24 months
Characterized by microcytic anemia, failure to thrive, fever, diarrhea, liver and spleen enlargement, and progressive pallor
Complications from iron overload include growth retardation, failure of sexual maturation, dilated cardiomy opathy, pericarditis, liver fibrosis and cirrhosis, diabetes, and parathyroid, thyroid, and pituitary insufficiency
Death often occurs in second or third decade due to cardiac complications
Untreated individuals exhibit severe failure to thrive, jaundice, increased skin pigmentation, liver and spleen enlargement, poor musculature, “chipmunk face,” skeletal deformities, and stunted growth and have a shortened life expectancy
♦
Beta thalassemia intermedia has m ilder phenotype and results from homozygosity or compound heterozygosity of alleles with reduced beta chain production; it may result from coinheritance of alpha and beta thalassemia
Variable presentation may include milder anemia, liver and spleen enlargement, moderate skeletal features, pallor, jaundice, cholelithiasis, osteopenia and osteoporosis, and extramedullary erythropoietic masses
There is an increased risk for iron overload due to increased absorption of iron in the intestine
Transfusions are performed as needed
♦
Beta thalassemia minor or thalassemia trait refers to a heterozygous carrier state. Carriers generally do not exhibit clinical symptoms other than possible mild anemia
Molecular Basis of Disease
♦
Beta thalassemia results from mutations in the HBB gene, which encodes for the beta hemoglo bin subunits
♦
The clinical presentation of beta thalassemia is dependent on the extent to which the genetic alteration disrupts beta chain production ranging from complete absence of production (i.e., β0 mutations) to “silent” or mild production disturbances (β+ mutations) and subsequently imbalance in the alpha to non-alpha globin chain ratio
♦
Normally, both alpha and beta globin chains are produced in roughly equal amounts. When beta globin synthesis is decreased, there are more free alpha chains. Free alpha chain s are very unstable and precipitate in the red cell. This results in ineffective erythropoiesis and anemia. Subsequently, there is a phenomenal increase in erythropoiesis with marrow expansion and persistence of erythropoiesis in the liver and spleen
♦
More than 200 genetic alterations hav e been described in the HBB gene, including:
Nucleotide substitutions
Deletions
Transcriptional mutations
RNA processing mutations
♦
Molecular analysis may be helpful in determining clinical phenotype and identifying at-risk relatives
Laboratory
♦
Red blood cell indices reveal microcytic anemia
♦
Peripheral blood smears show nucleated red blood cells, microcytosis, hypochromia, anisocytosis, and poikilocytosis
Carriers have reduced MCV and M CH, and RBC morphology is less severe than in affected individuals. Nucleated red blood cells are generally not seen in carriers
♦
Elevated iron
♦
Hyperplastic marrow with hyperplasia of red cell precursors
♦
Hemoglob in analysis shows reduced amounts of Hb A (normal adult hemoglobin) and increases in normal minor hemoglobins, such as Hb A2
♦
Molecular analysis is clinically available
Treatment
♦
Thalassemia major
Regular transfusions to correc t anemia, suppress erythropoiesis, and inhibit gastrointestinal absorption of iron
Iron chelation to prevent iron overload
Bone marrow transplantation from HLA-identical sib or cord blood transplantation from related donor
Ongoing surveillanc e is necessary to monitor effectiveness of transfusion and chelation therapy
•
Regular physical exams including growth and development assessment
•
Liver function tests; serum ferritin
•
Ophthalmologic, audiologic, and cardiac exams
•
Endocrine function and liver ultrasound
•
In adults, bone densitometry, serum AFP, and gallbladder echography are also assessed
♦
Thalassemia intermedia
Symptom atic therapy
Splenectomy
Folic acid supplementation
Sporadic red cell transfusions
Radiotherapy, transfusions, and hydroxyurea for treatment of extramedullary erythropoietic masses
Chelation therapy if iron overload develops
Sickle Cell Anemia
Chromosome and Gene Location
♦
11p15.5
Inheritance
♦
Autosomal recessive
Incidence
♦
1/500 African-Ame rican births
Clinical Manifestations
♦
Severe anemia
♦
Painful swelling and crisis due to vaso-occlusive episodes usually affecting the limbs, back, abdomen, and chest
♦
Pneumococcal sepsis
♦
Leg ulcerations
♦
Enlarged spleen
♦
Pallor
♦
Acute chest syndrome (e.g., the presence of infiltrate on chest radiograph, sometimes including lower respiratory tract symptoms, fever, and hypoxemia)
♦




Autosplenect omy (i.e., the reduction in spleen size due to continual splenic infarctions resulting from the filtration of abnormal red blood cells)
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
