INTRODUCTION
Viruses are etiologic factors in the development of several types of human tumors, including two of great significance worldwide—cervical cancer and liver cancer. At least 15–20% of all human tumors worldwide have a viral cause. The viruses that have been strongly associated with human cancers are listed in Table 43-1. They include human papillomaviruses (HPVs), Epstein-Barr virus (EBV), human herpesvirus 8, hepatitis B virus, hepatitis C virus, and two human retroviruses plus several candidate human cancer viruses. New cancer-associated viruses are being discovered by the use of molecular techniques. Many viruses can cause tumors in animals, either as a consequence of natural infection or after experimental inoculation.
| Virus Family | Virus | Human Cancer |
|---|---|---|
| Papillomaviridae | Human papillomaviruses | Genital tumors |
| Squamous cell carcinoma | ||
| Oropharyngeal carcinoma | ||
| Herpesviridae | Epstein-Barr virus | Nasopharyngeal carcinoma |
| Burkitt lymphoma | ||
| Hodgkin disease | ||
| B-cell lymphoma | ||
| Human herpesvirus 8 | Kaposi sarcoma | |
| Hepadnaviridae | Hepatitis B virus | Hepatocellular carcinoma |
| Polyomaviridae | Merkel cell virus | Merkel cell carcinoma |
| Retroviridae | Human T-lymphotropic virus | Adult T-cell leukemia |
| Human immunodeficiency virus | AIDS-related malignancies | |
| Flaviviridae | Hepatitis C virus | Hepatocellular carcinoma |
Animal viruses are studied to learn how a limited amount of genetic information (one or a few viral genes) can profoundly alter the growth behavior of cells, ultimately converting a normal cell into a neoplastic one. Such studies reveal insights into growth regulation in normal cells. Tumor viruses are agents that can produce tumors when they infect appropriate animals. Many studies are done using cultured animal cells rather than intact animals, because it is possible to analyze events at cellular and subcellular levels. In such cultured cells, tumor viruses can cause “transformation.” However, animal studies are essential to studying many steps in carcinogenesis, including complex interactions between virus and host and host responses to tumor formation.
Studies with RNA tumor viruses revealed the involvement of cellular oncogenes in neoplasia; DNA tumor viruses established a role for cellular tumor suppressor genes. These discoveries revolutionized cancer biology and provided the conceptual framework for the molecular basis of carcinogenesis.
GENERAL FEATURES OF VIRAL CARCINOGENESIS
Tenets of viral carcinogenesis are summarized in Table 43-2.
|
Like other viruses, tumor viruses are classified among different virus families according to the nucleic acid of their genome and the biophysical characteristics of their virions. Most recognized tumor viruses either have a DNA genome or generate a DNA provirus after infection of cells (hepatitis C virus is an exception).
DNA tumor viruses are classified among the papilloma-, polyoma-, adeno-, herpes-, hepadna-, and poxvirus groups. DNA tumor viruses encode viral oncoproteins that are important for viral replication but also affect cellular growth control pathways.
Most RNA tumor viruses belong to the retrovirus family. Retroviruses carry an RNA-directed polymerase (reverse transcriptase) that constructs a DNA copy of the RNA genome of the virus. The DNA copy (provirus) becomes integrated into the DNA of the infected host cell, and it is from this integrated DNA copy that all proteins of the virus are translated.
RNA tumor viruses are of two general types with respect to tumor induction. The highly oncogenic (direct-transforming) viruses carry an oncogene of cellular origin. The weakly oncogenic (slowly transforming) viruses do not contain an oncogene and induce leukemias after long incubation periods by indirect mechanisms. The two known cancer-causing retroviruses in humans act indirectly. Hepatitis C virus, a flavivirus, does not generate a provirus and appears to induce cancer indirectly.
Carcinogenesis is a multistep process; that is, multiple genetic changes must occur to convert a normal cell into a malignant one. Intermediate stages have been identified and designated by terms such as “immortalized,” “hyperplastic,” and “preneoplastic.” Tumors usually develop slowly over a long period of time. The natural history of human and animal cancers suggests a multistep process of cellular evolution, probably involving cellular genetic instability and repeated selection of rare cells with some selective growth advantage. The number of mutations underlying this process is estimated to range from five to eight. Observations suggest that activation of multiple cellular oncogenes and inactivation of tumor suppressor genes are involved in the evolution of tumors whether or not a virus is involved.
It appears that a tumor virus usually acts as a cofactor, providing only some of the steps required to generate malignant cells. Viruses are necessary—but not sufficient—for development of tumors with a viral etiology. Viruses often act as initiators of the neoplastic process and may do so by different mechanisms.
MOLECULAR MECHANISMS OF CARCINOGENSIS
“Oncogene” is the general term given to genes that are involved in cancer causation. Normal versions of these transforming genes are present in normal cells and have been designated proto-oncogenes.
The discovery of cellular oncogenes came from studies with acutely transforming retroviruses. It was found that normal cells contained highly related (but not identical) copies of various retrovirus transforming genes; cellular sequences had been captured and incorporated into the retrovirus genomes. Transduction of the cellular genes was probably an accident, as the presence of the cellular sequences is of no benefit to the viruses. Many other known cellular oncogenes that have not been segregated into retrovirus vectors have been detected using molecular methods.
Cellular oncogenes are partly responsible for the molecular basis of human cancer. They represent individual components of complicated pathways responsible for regulating cell proliferation, division, and differentiation and for maintaining the integrity of the genome. Incorrect expression of any component might interrupt that regulation, resulting in uncontrolled growth of cells (cancer). Examples exist of tyrosine-specific protein kinases (eg, src), growth factors (sis is similar to human platelet-derived growth factor, a potent mitogen for cells of connective tissue origin), mutated growth factor receptors (erb-B is a truncated epidermal growth factor receptor), GTP-binding proteins (Ha-ras), and nuclear transcription factors (myc, jun).
The molecular mechanisms responsible for activating a benign proto-oncogene and converting it into a cancer gene vary—but all involve genetic damage. The gene may be overexpressed, and a dosage effect of the overproduced oncogene product may be important in cellular growth changes. These mechanisms might result in constitutive activity (loss of normal regulation), so that the gene is expressed at the wrong time during the cell cycle or in inappropriate tissue types. Mutations might alter the carefully regulated interaction of a proto-oncogene protein with other proteins or nucleic acids. Insertion of a retroviral promoter adjacent to a cellular oncogene may result in enhanced expression of that gene (ie, “promoter-insertion oncogenesis”). Expression of a cellular gene also may be increased through the action of nearby viral “enhancer” sequences.
A second class of human cancer genes is involved in tumor development. These are the negative regulators of cell growth, tumor suppressor genes. They were identified because they form complexes with oncoproteins of certain DNA tumor viruses. The inactivation or functional loss of both alleles of such a gene is required for tumor formation—in contrast to the activation that occurs with cellular oncogenes. The prototype of this inhibitory class of genes is the retinoblastoma (Rb) gene. The Rb protein inhibits entry of cells into S phase by binding to key transcription factors that regulate expression of S-phase genes. The function of normal Rb protein is regulated by phosphorylation. The loss of Rb gene function is causally related to the development of retinoblastoma—a rare ocular tumor of children—and other human tumors.
Another crucial tumor suppressor gene is the p53 gene. It also blocks cell cycle progression; p53 acts as a transcription factor and regulates the synthesis of a protein that inhibits the function of certain cell cycle kinases. It also causes cells with DNA damage to undergo apoptosis. The loss of p53 function allows cells with damaged DNA to progress through the cell cycle, leading to the eventual accumulation of genetic mutations. The p53 gene is mutated in over half of all human cancers.
INTERACTIONS OF TUMOR VIRUSES WITH THEIR HOSTS
The pathogenesis of a viral infection and the response of the host are integral to understanding how cancer might arise from that background. The known tumor viruses establish long-term persistent infections in humans. Because of differences in individual genetic susceptibilities and host immune responses, levels of virus replication and tissue tropisms may vary among persons. Even though very few cells in the host may be infected at any given time, the chronicity of infection presents the long-term opportunity for a rare event to occur that allows survival of a cell with growth control mechanisms that are virus-modified.
Viruses that establish persistent infections must avoid detection and recognition by the immune system that would eliminate the infection. Different viral evasion strategies have been identified, including restricted expression of viral genes that makes infected cells nearly invisible to the host (EBV in B cells), infection of sites relatively inaccessible to immune responses (HPV in the epidermis), mutation of viral antigens that allows escape from antibody and T-cell recognition (human immunodeficiency virus [HIV]), modulation of host major histocompatibility complex class I molecules in infected cells (adenovirus, cytomegalovirus), inhibition of antigen processing (EBV), and infection and suppression of essential immune cells (HIV).
It is believed that host immune surveillance mechanisms usually eliminate the rare neoplastic cells that may arise in normal individuals infected with cancer viruses. However, if the host is immunosuppressed, cancer cells are more likely to proliferate and escape host immune control. Immunosuppressed organ transplant recipients and HIV-infected individuals are at increased risk of EBV-associated lymphomas and of HPV-related diseases. It is possible that variations in individual immune responses may contribute to susceptibility to virus-induced tumors in normal hosts.
Tumor viruses mediate changes in cell behavior by means of a limited amount of genetic information. There are two general patterns by which this is accomplished: The tumor virus introduces a new “transforming gene” into the cell (direct-acting), or the virus alters the expression of a preexisting cellular gene or genes (indirect-acting). In either case, the cell loses control of normal regulation of growth processes. DNA repair pathways are frequently affected, leading to genetic instability and a mutagenic phenotype.
Viruses usually do not behave as complete carcinogens. In addition to changes mediated by viral functions, other alterations are necessary to disable the multiple regulatory pathways and checkpoints in normal cells to allow a cell to become completely transformed. There is no single mode of transformation underlying viral carcinogenesis. At the molecular level, oncogenic mechanisms by human tumor viruses are very diverse.
Cellular transformation may be defined as a stable, heritable change in the growth control of cells in culture. No set of characteristics invariably distinguishes transformed cells from their normal counterparts. In practice, transformation is recognized by the cells’ acquisition of some growth property not exhibited by the parental cell type. Transformation to a malignant phenotype is recognized by tumor formation when transformed cells are injected into appropriate test animals.
Indirect-acting tumor viruses are not able to transform cells in culture.
At the cellular level, host cells are either permissive or nonpermissive for replication of a given virus. Permissive cells support viral growth and production of progeny virus; nonpermissive cells do not. Especially with the DNA viruses, permissive cells are often killed by virus replication and are not transformed unless the viral replicative cycle that results in death of the host cell is blocked in some way; nonpermissive cells may be transformed. However, there are situations in which DNA virus replication does not lyse the host cell and such cells may be transformed. Nevertheless, transformation is a rare event. A characteristic property of RNA tumor viruses is that they are not lethal for the cells in which they replicate. Cells that are permissive for one virus may be nonpermissive for another.
Not all cells from the natural host species are susceptible to viral replication or transformation or both. Most tumor viruses exhibit marked tissue specificity, a property that probably reflects the variable presence of surface receptors for the virus, the ability of the virus to cause disseminated versus local infections, or intracellular factors necessary for viral gene expression.
Some viruses are associated with a single tumor type, whereas others are linked to multiple tumor types. These differences reflect the tissue tropisms of the viruses.
The stable genetic change from a normal to a neoplastic cell generally requires the retention of viral genes in the cell. Often, but not always, this is accomplished by the integration of certain viral genes into the host cell genome. With DNA tumor viruses, a portion of the DNA of the viral genome may become integrated into the host cell chromosome. Sometimes, episomal copies of the viral genome are maintained in tumor cells. With retroviruses, the proviral DNA copy of the viral RNA is integrated in the host cell DNA. Genome RNA copies of hepatitis C virus that are not integrated are maintained in tumor cells.
In some viral systems, virus-transformed cells may release growth factors that affect the phenotype of neighboring uninfected cells, thereby contributing to tumor formation. It is also possible that as tumor cells collect genetic mutations during tumor growth, the need for the viral genes that drove tumor initiation may become unnecessary and viral markers will be lost from some cells.
RNA TUMOR VIRUSES
Hepatitis B virus (see Chapter 35), a member of the Hepadnaviridae family, is characterized by 42-nm spherical virions with a circular genome of double-stranded DNA (3.2 kbp). One strand of the DNA is incomplete and variable in length. Studies of the virus are hampered because it has not been grown in cell culture.
In addition to causing hepatitis, hepatitis B virus is a risk factor in the development of liver cancer in humans. Epidemiologic and laboratory studies have proved persistent infection with hepatitis B virus to be an important cause of chronic liver disease and the development of hepatocellular carcinoma. Hepatitis B virus infections occurring in adults are usually resolved, but primary infections in neonates and young children tend to become chronic in up to 90% of cases. It is these persistent hepatitis B virus infections established early in life that carry the highest risk of hepatocellular carcinoma later in life. The mechanism of oncogenesis remains obscure. Persistent viral infection leads to necrosis, inflammation, and liver regeneration that, over time, results in cirrhosis; hepatocellular carcinoma usually arises out of this background. The hepatitis B virus transactivator protein, X protein, is a potential viral oncoprotein. A dietary carcinogen, aflatoxin, may be a cofactor for hepatocellular carcinoma, especially in Africa and China.
The advent of an effective hepatitis B vaccine for the prevention of primary infection raised the possibility of prevention of hepatocellular carcinoma, particularly in areas of the world where infection with hepatitis B virus is hyperendemic (eg, Africa, China, Southeast Asia). Twenty years after the initiation of a universal hepatitis B vaccination program in Taiwan, chronic hepatitis B virus infection rates and liver cancer incidence rates were markedly reduced.
Woodchucks are an excellent model for hepatitis B virus infections of humans. A similar virus, woodchuck hepatitis virus, establishes chronic infections in both newborn and adult woodchucks, many of which develop hepatocellular carcinomas within a 3-year period.
Hepatitis C virus (see Chapter 35), a member of the Flaviviridae family, contains a genome of single-stranded RNA 9.4 kb in size. It appears that the majority of infections become persistent, even in adults. Chronic infection with hepatitis C virus leads to chronic inflammation and cirrhosis, and is also considered to be a causative factor in hepatocellular carcinoma. The development of hepatocellular carcinoma is likely mediated by a combination of virus and host specific mechanisms. There are over 170 million chronic carriers of hepatitis C virus, with 1-5% of these going on to develop hepatocellular carcinoma.
There are currently over 350 million people worldwide persistently infected with hepatitis B virus leading to over 500,000 deaths from hepatocellular cancer annually.
Retroviruses contain an RNA genome and an RNA-directed DNA polymerase (reverse transcriptase). RNA tumor viruses in this family mainly cause tumors of the reticuloendothelial and hematopoietic systems (leukemias, lymphomas) or of connective tissue (sarcomas).
Important properties of the retroviruses are listed in Table 43-3.
| Virion: Spherical, 80–110 nm in diameter, helical nucleoprotein within icosahedral capsid |
| Composition: RNA (2%), protein (about 60%), lipid (about 35%), carbohydrate (about 3%) |
| Genome: Single-stranded RNA, linear, positive-sense, 7–11 kb, diploid; may be defective; may carry oncogene |
| Proteins: Reverse transcriptase enzyme contained inside virions |
| Envelope: Present |
| Replication: Reverse transcriptase makes DNA copy from genomic RNA; DNA (provirus) integrates into cellular chromosome; provirus is template for viral RNA |
| Maturation: Virions bud from plasma membrane |
Outstanding characteristics: Infections do not kill cells May transduce cellular oncogenes; may activate expression of cell genes Proviruses remain permanently associated with cells and are frequently not expressed Many members are tumor viruses |
The retrovirus genome consists of two identical subunits of single-stranded, positive-sense RNA, each 7–11 kb in size. The reverse transcriptase contained in virus particles is essential for viral replication.
Retrovirus particles contain the helical ribonucleoprotein within an icosahedral capsid that is surrounded by an outer membrane (envelope) containing glycoprotein and lipid. Type- or subgroup-specific antigens are associated with the glycoproteins in the viral envelope, which are encoded by the env gene; group-specific antigens are associated with the virion core, which are encoded by the gag gene.
Three morphologic classes of extracellular retrovirus particles—as well as an intracellular form—are recognized, based on electron microscopy. They reflect slightly different processes of morphogenesis by different retroviruses. Examples of each are shown in Figure 43-1.
FIGURE 43-1
Comparative morphology of types A, B, C, and D retroviruses. A: Intracytoplasmic type A particles (representing immature precursor of budding type B virus). B: Budding type B virus. C: Mature, extracellular type B virus. D: Lack of morphologically recognizable intracytoplasmic form for type C virus. E: Budding type C virus. F: Mature, extracellular type C virus. G: Intracytoplasmic type A particle (representing immature precursor form of type D virus). H: Budding type D virus. I: Mature, extracellular type D virus. All micrographs are approximately 87,000×. Thin sections were double-stained with uranyl acetate and lead citrate. (Courtesy of D Fine and M Gonda.)
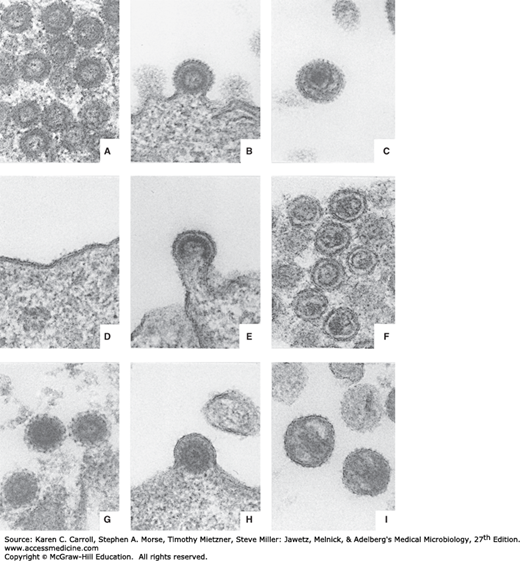
Type A particles occur only intracellularly and appear to be noninfectious. Intracytoplasmic type A particles, 75 nm in diameter, are precursors of extracellular type B viruses, whereas intracisternal type A particles, 60–90 nm in diameter, are unknown entities. Type B viruses are 100–130 nm in diameter and contain an eccentric nucleoid. The prototype of this group is the mouse mammary tumor virus, which occurs in “high mammary cancer” strains of inbred mice and is found in particularly large amounts in lactating mammary tissue and milk. It is readily transferred to suckling mice, in whom the incidence of subsequent development of adenocarcinoma of the breast is high. The type C viruses represent the largest group of retroviruses. The particles are 90–110 nm in diameter, and the electron-dense nucleoids are centrally located. The type C viruses may exist as exogenous or endogenous entities (see as follows). The lentiviruses are also type C viruses. Finally, the type D retroviruses are poorly characterized. The particles are 100–120 nm in diameter, contain an eccentric nucleoid, and exhibit surface spikes shorter than those on type B particles.
The Retroviridae family is divided into seven genera: Alpharetrovirus (which contains avian leukosis and sarcoma viruses), Betaretrovirus (mouse mammary tumor virus), Gammaretrovirus (mammalian leukemia and sarcoma viruses), Deltaretrovirus (human T-lymphotropic viruses [HTLV] and bovine leukemia virus), Epsilonretrovirus (fish viruses), Spumavirus (which contains viruses able to cause “foamy” degeneration of inoculated cells but which are not associated with any known disease process), and Lentivirus (which encompasses agents able to cause chronic infections with slowly progressive neurologic impairment, including HIV; see Chapter 44).
Retroviruses can be organized in various ways depending on their morphologic, biologic, and genetic properties. Differences in genome sequences and natural host range are frequently used, but antigenic properties are not. Retroviruses may be grouped morphologically (types B, C, and D); the vast majority of isolates display type C characteristics.
Retroviruses have been isolated from virtually all vertebrate species. Natural infections by a given virus are usually limited to a single species, though infections across species barriers may occur. Group-specific antigenic determinants on the major internal (core) protein are shared by viruses from the same host species. All mammalian viruses are more closely related to one another than to those from avian species.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


