Fig. 1.1
Genetic map of the human and mouse MHCs. (a) Schematic map of human MHC on chromosome 6. Illustrations are not drawn to scale. The centromere (circle) and major HLA are indicated in order. The number of distinct proteins (alleles) encoded by each human MHC gene is indicated under each locus. (b) Based on the DRB1 allele type, human MHC haplotypes fall into four groups—DR51, DR52, DR53, and DR1/10/8—that vary in DRB gene number. The serological specificities encoded by each DRB gene are provided underneath each locus (loci are indicated by boxes). The number of alleles encoded by each DRB gene is indicated. (a, Data from IMGT/HLA database, release 3.19; http://www.ebi.ac.uk/imgt/hla/stats.html [1, 2]; b, Data from IMGT/HLA database, January 2015; http://www.ebi.ac.uk/imgt/hla/stats.html [1, 2])
The HLA-DR genomic region contains one functional gene for the α-chain (DRA), which pairs with one or two functional genes for the β-chain (DRB1, DRB3, DRB4, DRB5) resulting in expression of the DR51, DR52, or DR53 antigens depending on the HLA-DRB1 allele type (Fig. 1.1b) [1, 2]. The HLA class III region, between the genes for class I and class II on chromosome 6, contains many immune genes involved in immune function including complement and cytokine genes (Fig. 1.1a).
Structurally, the HLA class I molecule is a heterodimer consisting of a heavy α chain that is bound noncovalently to β2-microglobulin (β2m) light chain, a non-MHC gene (located in chromosome 15) (Fig. 1.2). The HLA class II molecule is composed of two transmembrane glycoprotein chains, α (encoded by DRA, DQA1, or DPA1) and β (encoded by DRB, DQB1, or DPB1). Each chain has two domains, and the α1 and β1 domains of class II molecules form the peptide-binding cleft. The pockets located in the groove accommodate distinct peptides with specific characteristics (Fig. 1.2). Therefore, each HLA binds different peptides.
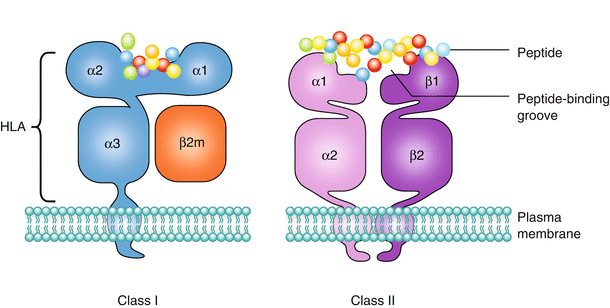
Fig. 1.2
Structure of HLA molecules. HLA class I and class II molecules present peptide antigens to CD8+ and CD4+ T cells, respectively. The HLA class I molecule is a heterodimer of a membrane-spanning heavy α chain (encoded by HLA-A, -B, or –C gene) bound noncovalently to a non-MHC gene (located in chromosome 15)–encoded light β2-microglobulin (β2m) chain, which does not span the membrane. The α chain folds into three domains: α1, α2, and α3. The α1 and α2 domains form the antigen-binding groove. The HLA class II molecule is composed of two transmembrane glycoprotein chains, α (encoded by DRA, DQA1 or DPA1) and β (encoded by DRB1, DQB1 or DPB1). Each chain has two domains, and the two chains together form a compact four-domain structure similar to that of a HLA class I molecule. The α2 and β2 domains, like the α3- and β2-microglobulin domains of the HLA class I molecule, have amino acid sequence and structural similarities to IgC domains. The α1 and β1 domains of class II molecules form the peptide-binding cleft. The major difference between class I and class II is that the ends of the peptide-binding groove are more open in HLA class II molecules than in HLA class I molecules. As a result, the HLA class I can accommodate only short peptides (~9 amino acids long) and the ends of the peptide are substantially buried within the class I molecule. In contrast, the class II groove has open ends and can accommodate longer peptides (12–20 amino acids long)
The T-cell receptor recognizes self-HLA molecules containing a peptide fragment to elicit an immune response. In the setting of allotransplantation, the patient’s T-cell receptors can recognize intact donor HLA molecules through a pathway called “direct” allorecognition as described later.
1.1.2 HLA Gene Polymorphism, Haplotypes, and Inheritance
The most notable feature of the human MHC is the remarkable degree of polymorphism. To date, over 9400 distinct HLA class I alleles and 3000 class II alleles have been recognized [1]. The polymorphic regions are principally localized to the amino terminal region of these molecules, which bind peptides and interact with T-cell receptors. Although the high degree of HLA polymorphism is necessary for enhancing the diversity in the repertoire of HLA-bound peptides to combat pathogens, it creates a significant barrier for transplantation of histoincompatible tissues and organs between individuals.
The collection of MHC alleles present on each parental chromosome is called an HLA haplotype. HLAs are inherited in Mendelian fashion, which means that each parental chromosome 6 provides a haplotype or linked set of MHC genes to the offspring. A child is a one-haplotype match to each parent unless recombination, or a “crossover,” between genes of the parental haplotype occurs. Statistically, there is a 25 % chance that siblings will share the same parental haplotypes (HLA-identical), a 50 % chance they will share one haplotype (one-haplotype match), and a 25 % chance that neither haplotype will be the same (zero-haplotype match) (Fig. 1.3).
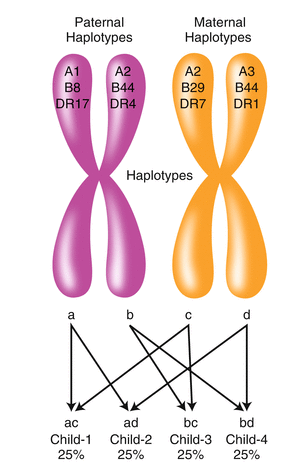
Fig. 1.3
HLA haplotype segregation in a family. Each child inherits one HLA haplotype from each parent. Because each parent has two different haplotypes (paternal = ab and maternal = cd), four different haplotypic combinations are possible in the offspring (ac, ad, bc, bd). Therefore, a child has a 25 % chance of having an HLA-identical– or zero-haplotype–matched sibling donor, and a 50 % chance of having a one-haplotype–matched sibling donor. All children have one-haplotype matched to each parent unless recombination has occurred
Box 1.1
The HLA region demonstrates strong linkage disequilibrium across HLA-A, -B, -C, -DR, -DQ, and -DP alleles. Linkage disequilibrium is a phenomenon in which alleles at adjacent HLA loci are inherited together more often than would be expected by chance. Existing data suggest that positive selection is operating on the haplotype and that the linked loci confer a particular selective advantage for the host.
1.1.3 HLA Matching for Solid Organ Transplantation
Prior to transplant, recipients and donors are HLA-typed and mismatched antigens are identified. It is well established that renal transplants from HLA-identical sibling donors survive 50 % longer on average than transplants from HLA-mismatched living donors [3]. Similarly, kidney grafts from deceased donors that have no HLA-A, -B, or -DR locus antigen mismatches survive longer than grafts from donors carrying HLA mismatches. In order to increase the number of HLA-matched transplants, the degree of HLA compatibility has been incorporated into deceased donor kidney allocation systems in many countries. However, given the extensive polymorphism of the HLA system, grafts carrying similar HLA genotypes from unrelated donors are uncommon. HLA matching is, therefore, not considered in the allocation of most solid organ transplants except for national sharing of HLA-matched renal allografts for highly sensitized recipients and HLA-DR matching allocation of locally procured kidneys.
1.1.4 Cellular Responses to HLA Alloantigens
Allorecognition is the activation of the transplant recipient’s adaptive immune response to mismatched donor histocompatibility antigens following transplant [4, 5]. Activation of the recipient’s CD4+ T lymphocytes is a pivotal step in the initiation of the immune response to alloantigens following transplantation leading to downstream activation of cytotoxic CD8+ T lymphocytes and antibody-producing B cells. Evidence supports three mechanisms of allorecognition: the direct, indirect, and semidirect pathways (Fig. 1.4). These pathways of allorecognition can occur independently or simultaneously and are associated with graft pathology and transplant outcome.
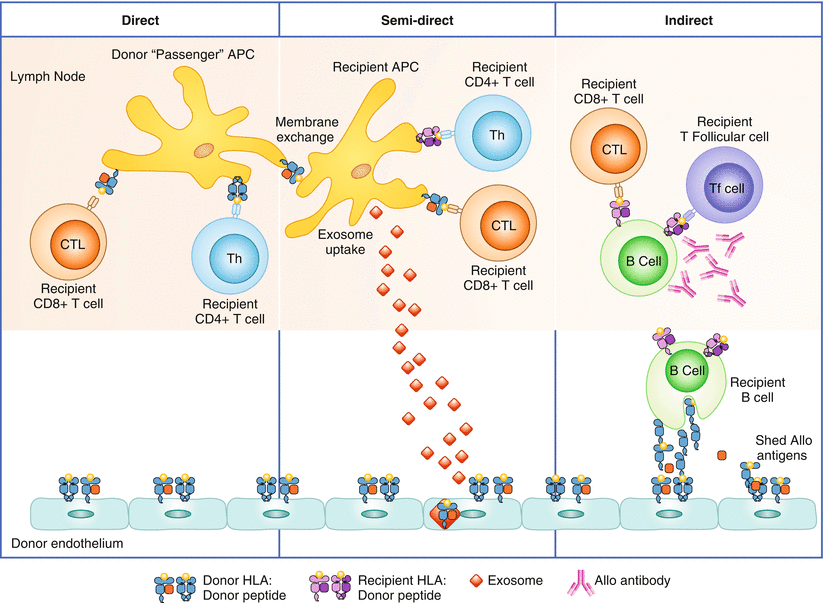
Fig. 1.4
Alloimmune responses occur through direct, indirect, and semidirect recognition. The direct pathway involves presentation of allogeneic MHC class I and II antigens on donor APCs to recipient CD4+ and cytotoxic CD8+ T cells (CTL) and is believed to be the primary mechanism of acute rejection. The indirect pathway involves processing the donor alloantigens by recipient B cells and presentation to recipient T-follicular cells and CTL. Alloantibodies are generated by interaction of alloreactive B cells with CD4+ T cells, and can lead to AMR. The indirect recognition pathway is considered a major pathway mediating chronic rejection. In the semidirect pathway, intact donor HLA class I:peptide complexes are presented on the DC of the recipient (through either membrane exchange or exosome uptake) to recipient CD8+ T cells. Simultaneously, processed donor peptide is presented in the context of the recipient’s HLA class II to the recipient’s CD4+ T cells. The recipient’s T-helper cells, activated by the indirect pathway, can then provide “help” to the recipient’s CTL, activated by the direct pathway. Blue indicates donor HLA class I and II. Purple indicates recipient HLA class I and II
The direct pathway is the activation of the transplant recipient’s CD4+ and CD8+ T cells by HLA:peptide complexes on the surface of donor cells that are transplanted as passengers with the organ. The donor’s antigen presenting cells (APCs), expressing foreign HLA, migrate to the secondary lymph nodes of the recipient and present donor antigens to the recipient’s CD4+ T cells. The strength of the immune response elicited by the direct allorecognition pathway correlates to the high frequency of recipient allogeneic T cells that become activated during the first few weeks following transplant, mediating acute rejection. CD4+ T cells activated through the direct pathway are capable of providing help to effector CD8+ T cells, therefore promoting cell-mediated rejection of the transplanted organ (Fig. 1.4).
In contrast to the direct allorecognition pathway, indirect allorecognition is the activation of the transplant recipient’s CD4+ T cells by alloantigen that is processed and presented in the context of the recipient’s HLA. Donor antigens, shed by the grafted organ, are processed and presented in the context of self-restricted HLA class II by the recipient’s B cells. The recipient’s follicular helper CD4+ T cells are then activated to provide help, leading to the generation of alloreactive CD8+ effector T cells and antibody-producing B cells (Fig. 1.4). The immune response engendered by this pathway can incite cell-mediated or antibody-mediated rejection and is credited with driving chronic rejection. Also, owing to the lower frequency of T cells with indirect allospecificity, and requirements for antigen processing, the indirect pathway is physiodynamically slower than the response to presentation through the direct pathway. It usually takes 14 days for de novo donor-specific antibodies (DSAs) to develop after transplantation.
The semidirect pathway of allorecognition is presented as a hypothesis to describe events of apparent overlap between the direct and the indirect pathways from animal models of transplant rejection indicating that indirectly activated allospecific CD4+ T cells can provide help to directly activated allospecific CD8+ T cells [6]. The mechanism underling the phenomenon of semidirect allorecognition likely lies in the exchange of membrane proteins between immune cells. After transplantation, the recipient’s dendritic cells (DCs) acquire intact donor HLA class I:peptide complexes from donor passenger DC or endothelial cells either through membrane exchange or by uptake of exosomes, or vesicles, containing the antigen that are shed from donor tissue. The recipient’s DC then bears intact donor HLA class I molecules as well as recipient HLA class II molecules, and is capable of stimulating the recipient’s CD4+ and CD8+ T cells via the indirect and direct pathways (Fig. 1.4).
Box 1.2
Common in vitro assays used to measure direct and indirect alloreactivity include the mixed lymphocyte culture (MLR), cytotoxic precursor T-cell assay, cytokine ELISPOT, or flow cytometry. To measure alloactivation via the direct allorecognition pathway, recipient T cells are cocultured with irradiated/inactivated donor APCs. For the indirect allorecognition pathway, recipient T cells are cocultured with autologous APCs pulsed with donor cellular protein fragments, or synthetic peptides.
1.1.5 HLA Typing
HLA antigens were initially defined using serological microlymphocytotoxicity techniques that used a battery of carefully selected antisera to recognize distinct HLAs on the surface of lymphocytes. During the past 20 years, more precise DNA-based typing techniques have replaced serological methods in clinical applications. The three basic DNA-based HLA typing techniques used in conjunction with polymerase chain reaction (PCR) are the reverse sequence-specific oligonucleotide (rSSO) probe hybridization method, sequence-specific primer (SSP) directed amplification method, and sequencing-based typing (SBT) method. The basic premise for these assays is that the HLA genes are selectively amplified by the PCR followed by detection of the specific polymorphisms by gel electrophoretic mobility (SSP) (Fig. 1.5a), direct DNA sequencing (SBT), or hybridization with sequence-specific oligonucleotide probes (rSSO) (Fig. 1.5b).
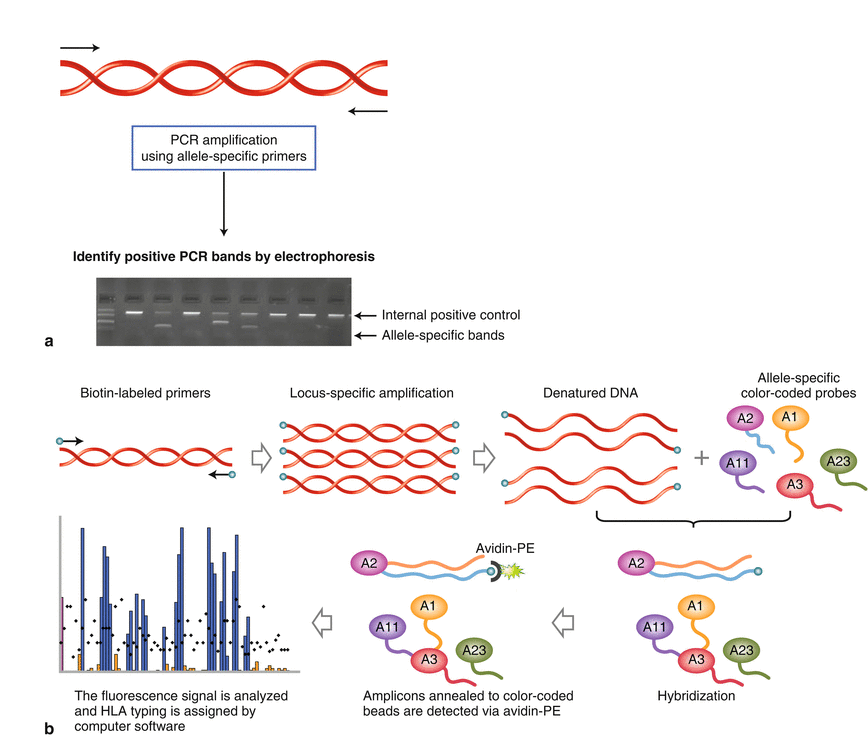
Fig. 1.5
Molecular HLA typing. (a) HLA typing by SSP. Each PCR well contains a unique set of primers that are designed to have perfect matches with a single allele or group of alleles and produce a product with a particular known size. A picture of an agarose gel electrophoresis shows the pattern of allele-/group-specific PCR products corresponding to amplification of HLA class I or class II genes. Under strictly controlled PCR conditions, perfectly matched primer pairs result in the amplification of target sequences and an identifiable band on an agarose gel (i.e., a positive reaction) whereas mismatched primer pairs do not amplify (i.e., a negative reaction). Each PCR reaction also includes a positive internal control primer pair that amplifies a conserved gene segment (i.e., human β-globulin gene), which is present in all human DNA samples and is used to verify the integrity of the PCR reaction. (b) The reverse single-nucleotide probe hybridization (SSOP) method using Luminex bead arrays. DNA samples are PCR-amplified using biotin-conjugated locus-specific primers. The PCR products are denatured and hybridized with probe arrays of Luminex color-coded polystyrene beads. Amplicons annealed to the polystyrene beads are detected via streptavidin phycoerythrin (PE) chemistry. Fluorescence signals of both PE and color-coded beads are detected by using the Luminex 100 flow-based instrument. The combined data are analyzed and HLA typing is assigned by a software program
Development of rapid, high-throughput sequencing and PCR assays for HLA typing have allowed for a more accurate definition of recipient and donor HLA genotypes. High-resolution HLA typing, which is achieved by SBT, allows for differentiation of unique epitopes to which a recipient may make specific antibodies. One major limitation of SBT is that it often results in ambiguous typing due to the sharing of nucleotide sequences across the specific exons interrogated and additional testing is required to report the allele-level typing results. Implementation of next-generation sequencing (NGS) technologies should eliminate the problem of HLA genotyping ambiguities. The NGS technology enables parallel sequencing of billions of PCR-amplified DNA fragments. Many groups have developed long-range PCR strategies to amplify the full length of the HLA genes, followed by fragmentation of the amplified DNA, and sequencing by NGS. Coupling the NGS strategy with sophisticated software algorithms to perform the assembly of the sequenced fragments provides in-phase, full-length allele-level typing.
1.2 Role of HLA Antibodies in Transplantation
Sensitization to non-self HLA through pregnancies, prior transplantation, and/or blood transfusions increases the risk of allograft rejection and limits the patient’s access to organ transplantation. The Organ Procurement Transplant Network data indicate as many as 30 % of patients waiting for transplantation are presensitized to HLA antigens. Circulating DSAs in a transplant candidate may damage the graft to varying degrees depending on the DSA titer, specificity, and level of HLAs expression on the graft. High-titered pre-transplant DSA directed against HLA class I antigens can cause catastrophic hyperacute rejection and immediate graft loss, whereas high-titer class II DSAs mediate graft rejection 2–4 days after transplant, upon re-expression of HLA class II antigens on the endothelium of the allograft. In contrast, pre-transplant DSAs of low titer are often associated with development of acute antibody-mediated rejection (AMR) during the first 3 months after transplantation. If left untreated, patients with AMR are at risk of graft loss and/or markedly shortened overall graft survival time. Patients producing de novo anti-HLA antibodies against their donor following transplantation are also at increased risk of graft failure unless their response can be controlled or abrogated. The effect of post-transplant DSAs on different transplanted organs may differ in acute severity, in the specific pathological lesions, and ultimately, the degree of damage they cause to the organ, but evidence is rapidly accruing that shows these antibodies can damage any transplanted organ including kidney, heart, lung, intestine, and liver. Standard treatment for AMR consists of repeated plasmapheresis, immunoadsorption, intravenous immune globulin (IVIG), splenectomy, and eculizumab. Various combinations of these therapeutic modalities have been successfully used to treat AMR and improve outcomes in some patients; however, in many cases, interventions for AMRs are not always effective.
1.2.1 Antibody Biology
Human immunoglobulins are divided into 5 isotypes (IgM, IgD, IgG, IgE, and IgA). IgG and IgA are further subdivided into several subclasses each (IgG1, IgG2, IgG3, and IgG4; and IgA1 and IgA2). Antibody isotype and subclass are important determinants of both affinity for antigen and capacity to trigger immune effector functions. Antibody structure, complement-fixing capacity, and affinity for Fc receptors (FcγRs) is illustrated in Fig. 1.6. IgG is the predominant isotype in circulation, and although antibodies to HLA may be IgM, IgA, or IgG, clinical studies to date have concluded that IgG to donor HLA is most clinically relevant, although recent evidence points to a pathogenic effect of persistent IgM.
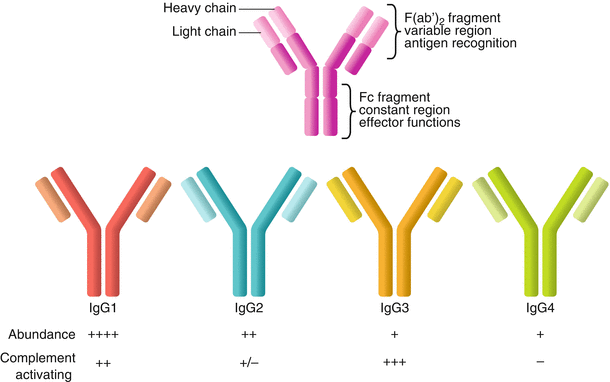
Fig. 1.6
Antibody structure and biology. Human immunoglobulin is composed of a light chain and a heavy chain, each of which contains a constant region and a variable region. The variable regions of the light and heavy chains combine to form the three-dimensional antigen-binding region. The constant region of the heavy chain forms the Fc fragment, which interacts with immune effector systems, including complement and innate immune cell Fc receptors. The predominant isotype in circulation is IgG, which consists of four subclasses—IgG1, IgG2, IgG3, and IgG4—numbered in order of their relative abundance in circulation. Each subclass has different constant regions and, therefore, a different capacity to elicit Fc-mediated functions, including activating of complement
1.2.2 Antibody Effector Functions
Recognition of antigen by the F(ab’)2 fragment elicits crosslinking of the target HLA and triggers activation of intracellular signaling cascades, described later (see Sect. 1.2.8). In addition, antibodies carry out their canonical effector functions through the Fc fragment, which bridges innate effector systems with adaptive immunity. Antibodies can activate the classical complement cascade to trigger production of soluble and cell-bound inflammatory mediators and can interact with receptors on myeloid and innate lymphoid cells to mediate cellular functions [7]. Both of these functions are reflected in the diagnostic criteria of AMR in kidney and heart allografts, manifesting as C4d deposition in the microvasculature and intravascular macrophage (CD68) or neutrophil infiltration in the allograft. In the following section, we briefly review the biology of the complement system and discuss the implications and limitations to the current use of markers of these processes in the pathological diagnosis of AMR.
1.2.3 Complement
With the recognition that antibodies to donor HLA can cause rejection in renal allografts came the need for accurate pathological markers of AMR. Detection of classical complement activation in renal allografts in regraft patients. In recipients with circulating antibodies and intense C3d and C4d deposition in peritubular capillaries was first reported in the early 1990s. The current diagnostic criteria for AMR in cardiac, renal, and pancreas allografts include immunohistochemical staining for C4d, as a pathological marker of activation of the complement cascade in the microvasculature. Although microvascular C4d alone is not a specific indicator of AMR, in conjunction with other pathological evidence of antibody-mediated injury, it has important utility in kidney, heart, and pancreas allografts. However, in other solid organs, including lung, liver, and small bowel, the clinical utility of C4d is unclear and currently no consensus criteria for the pathologic diagnosis of AMR exist. In order to understand why C4d is pathologically significant, why it may not always specifically indicate rejection, and why it may have variable utility among organs, it is necessary to briefly review the complement cascade.
1.2.3.1 What Is C4d?
Complement is an ancient system of innate immunity composed of circulating plasma proteins. Complement becomes activated at sites of inflammation to tag pathogens or dysfunctional host cells for destruction by facilitating opsonization, promoting recruitment of immune effector cells, and causing direct lysis of gram-negative bacteria, fungi, enveloped viruses, and antibody-coated host cells. Inactive under quiescent conditions, complement proteins include zymogens, which are activated upon cleavage and, through a series of catalytic events, generate inflammatory split products [8].
Complement activation proceeds stepwise from recognition of target surfaces, which triggers activation of catalytic complement proteins that successively cleave their targets (Fig. 1.7). The peptide products of cleavage form active convertases that cleave other complement proteins or inflammatory mediators called anaphylatoxins and opsonins. Terminal complement activation may cause lysis of the target cell, a phenomenon called complement-dependent cytotoxicity (CDC). However, host cells express a variety of cell surface complement regulatory receptors, and several complement inhibitory proteins are also present in the circulation. At each step, these soluble and membrane-bound host regulatory proteins degrade active complement components to restrict the magnitude, location, and duration of complement-mediated damage. For complement activation, particularly lysis, to occur at the surface of the host cell, it must exceed the threshold set by negative regulation by endogenous complement inhibitors. CDC resulting in cell death is, therefore, thought to be an exceptional event observed under conditions of extreme complement activation, such as during hyperacute rejection.
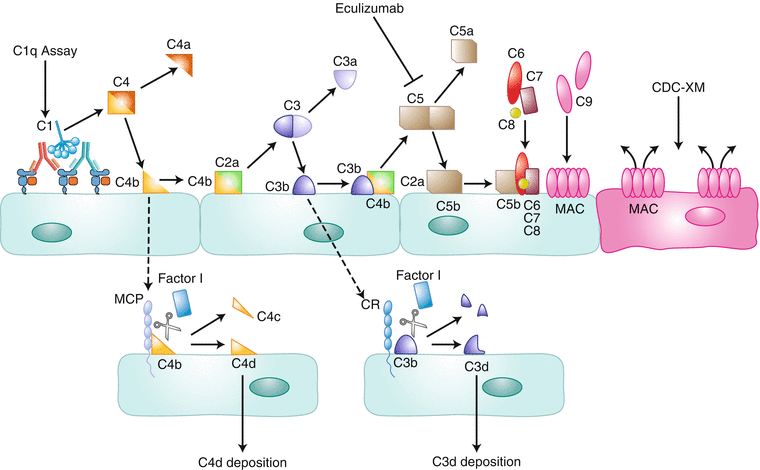
Fig. 1.7
The classical complement cascade. Antibody binding to antigen, in this case, to HLA class I on donor endothelial cells, in close proximity permits binding of the C1 complex and initiation of the classical complement cascade. The C1q assay measures the binding of HLA antibodies to C1q (a component of the C1 complex) on single-antigen beads. The C1 complex cleaves C4 to generate C4b. Catalytic products such as C4b can go on to form convertases and soluble mediators that perpetuate and amplify the complement cascade (solid arrows) or can be targeted by endogenous inhibitors to inactivate them (dashed arrows). For example, C4b either combines with C2a to form a catalytically active complex to cleave C3 or is degraded by complement regulatory proteins and inhibitors such as MCP and Factor I (dashed line), to generate C4d. C4d remains covalently linked to the cell membrane and is a marker of early complement activation. Cleavage of C3 generates the inflammatory product C3a and the cell surface–bound C3b. Like C4b, C3b can either be degraded by inhibitory proteins (complement receptors 1–3 and Factor I) to yield C3d or form a catalytic complex, with the C4b/C2a complex, that acts on C5. When C5 is cleaved, soluble C5a, a potent anaphylatoxin, and C5b are generated. Eculizumab is an anti-C5 antibody that binds to C5 with very high affinity and prevents its cleavage. C5b recruits several other complement proteins to the cell surface and ultimately promotes the formation of the MAC. MAC forms pores in the cell membrane and, at high levels, can cause lysis of the cell. This outcome is measured in the CDC-XM assay (Fig. 1.8)
Three pathways of complement are defined by the distinct mechanisms of initiation, although ultimately these pathways converge on the same downstream actors. The alternative complement pathway is activated directly at the cell surface of pathogens by deposition of spontaneously generated C3b and advances to terminal complement activation owing to the lack of inhibitory molecules on microbial surfaces. The lectin pathway also targets pathogens through specific recognition of microbial carbohydrate. Lastly, the classical pathway is activated by antibody bound to the surface of target cells, linking complement to adaptive immunity. Here, antibody is recognized by complement C1 complex that binds to the Fc region of certain isotypes and subclasses of immunoglobulin. It is this pathway that is thought to lie at the heart of HLA antibody–mediated allograft injury and rejection.
The classical complement pathway is initiated by IgM or certain subclasses of IgG binding to antigens on target cells, which are recognized by the C1q subunit of the C1 complex. The globular heads of C1q bridge the proximal Fc tails of antigen-complexed antibody. The capacity of an antibody to bind to C1q and elicit complement activation is dependent upon several independent factors, including antibody subclass, affinity, titer, and glycosylation. Binding of C1q to HLA antibodies can be detected in vitro using C1qScreen assay (Fig. 1.7). DSAs that bind C1q in this assay are predicted to activate the complement cascade (e.g., distinguishing the effective complement activator IgG3 from the relatively inactive IgG4). To date, the reports of the prognostic value of this assay have been somewhat variable, and more work is needed to elucidate its utility in predicting outcome.
Box 1.3
Complement C1 complex binding to antibody through C1q induces a conformation change in the catalytic subunit C1r. C1r then cleaves C1s to activate its serine protease function. Activated C1s is capable of cleaving C4 into C4a and C4b, the latter of which covalently attaches to the target surface near C1. Whereas C4a remains soluble and acts as an anaphylatoxin, C4b can either complex with a split product of C2 (C2a) generated by C1s to form an active convertase or be disabled by complement regulatory proteins. Uncleaved C4b in complex with C2a forms the C3 convertase, which cleaves C3 into soluble inflammatory C3a, and the immobilized opsonin C3b, which joins with C4bC2a to form the C5 convertase. Catalysis of C5 by this complex causes terminal complement activation, generating the potent anaphylatoxin C5a and the peptide C5b. C5b initiates formation of the MAC by recruiting C6, C7, C8, and C9. The MAC forms a pore that penetrates the target cell membrane and, at high concentrations, may cause lysis, a final outcome that is measured in the complement-dependent cytotoxicity crossmatch (CDC-XM) assay.
C1q:antibody binding triggers a complex cascade of cleavage events, producing soluble inflammatory mediators such as C3a and C5a, and cell surface–associated products including the membrane attack complex (MAC). The MAC forms a pore that penetrates the target cell membrane and may cause lysis. The potential for HLA antibodies to damage prospective donor cells through MAC formation is measured in the clinical test called complement-dependent cytotoxicity crossmatch (CDC-XM), discussed in more detail later (see Sect. 1.2.4). Notably, C5 cleavage is inhibited by the drug eculizumab, preventing production of C5a and formation of MAC.
Several complement split products remain covalently linked to the target cell membrane and can be detected immunohistologically. C4d is thus far the most useful of these for the diagnosis of AMR. It is generated from degradation of C4b by negative regulators (C4b-binding protein, complement receptor 1 [CR1], membrane cofactor protein [MCP, CD46], or Factor I), which release a C4c peptide and reveal the small C4d split product. C4d remains covalently linked to the cell membrane and has a longer half-life than C4b because of this turnover mechanism. Its immunohistological detection in the microvasculature indicates early activation of the complement cascade, specifically marking cleavage of C4. C3b is similarly regulated by inhibitory proteins, and its degradation is marked at the cell surface by C3d. In vitro assays measuring C3d generation by HLA antibodies in vitro may have added utility compared with C1qScreen, because C3d is much further downstream than C1q and this assay is functionally dependent upon physiological activation of complement (Fig. 1.7).
1.2.3.2 Why Might C4d Not Be Specific?
It is important to note that C4d is relatively far upstream in the classical complement cascade. Therefore, detection of C4d does not guarantee downstream terminal complement activation that leads to anaphylatoxin production or cell lysis (MAC). Indeed many regulatory checkpoints downstream of C4 prevent this in most cases, and endothelial cells are quite resistant to complement-dependent lysis in vitro. Interestingly, in C4d-positive cardiac biopsies, concurrent expression of the complement inhibitors decay accelerating factor (DAF), CD55, and CD59 on endothelium were increased in patients without allograft dysfunction but absent in those with impairment [9, 10]. These and other results suggest that, despite evidence of C4d deposition and early complement activation, endogenous complement inhibitory proteins may protect the allograft from damage during AMR. It has been suggested that immunohistological detection of C3d, which is counterintuitively downstream of C4d (Fig. 1.7), might be a superior indicator of more advanced complement activation [10].
In addition, clinically relevant HLA antibodies are of the IgG isotype. Blood group disparities are recognized by natural IgM, a potent activator of the classical complement cascade. IgM does not require high concentrations of antibody, because each molecule of IgM has multiple C1q-binding sites. C4d is very often detected in ABO-incompatible (ABOi) allografts without other evidence of rejection or graft dysfunction (discussed later in Sect. 1.3.4), and thus C4d alone is not a reliable indicator of HLA antibody-mediated graft injury or complement-induced damage. In addition, C4d is a possible product of both the classical (antibody-initiated) pathway and the lectin pathway, which also acts through C4. In contrast, the alternative pathway bypasses C4, as it relies on spontaneous C3 cleavage and C3b deposition on microbial surfaces. One report showed evidence for lectin pathway activation in C4d-positive renal allografts, based on simultaneous detection of the lectin pathway–specific molecule H-ficolin, suggesting that C4d might not be specific for antibody-dependent classical pathway activation in this setting [11].
Finally, experimental evidence indicates that HLA antibodies cause graft injury by additional complement-independent mechanisms. Indeed, the diagnostic criteria for AMR in renal and cardiac transplantation have recently been expanded to include recognition of C4d-negative AMR, based on clinical observations and molecular diagnostic studies showing AMR mechanisms involving endothelial injury and dysfunction in the absence of C4d deposition.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


