Overview
Hematopathology includes both diseases of red blood cells and diseases of white blood cells, as well as coagulation disorders. The first part of this chapter will discuss diseases of red blood cells, including general features of anemia, microcytic anemia, macrocytic anemia, and other anemias associated with decreased production, general features of hemolytic anemias, antibody-mediated destruction of red blood cells and other external causes of red blood cell destruction, hereditary causes of increased red blood cell destruction, and polycythemia. The second part of the chapter will discuss diseases of white blood cells and coagulation disorders.
Anemia
Overview: A shortage of red blood cells is referred to as anemia. There are three mechanisms by which the body becomes anemic: blood loss (either acute or chronic), decreased production of red blood cells, and increased destruction of red blood cells (i.e., hemolysis). These anemias can be categorized in two general ways: by mechanism (as outlined above), and by morphology, such as by mean corpuscular volume (MCV). To evaluate an anemia, the initial step is classification based upon the MCV, the reticulocyte count, and the blood smear. A high reticulocyte count indicates that the bone marrow is responding to the anemia by producing red blood cells. A low reticulocyte count indicates a production problem. The laboratory measures red cell indices based upon the size of the red blood cell and the amount of hemoglobin (Hb) per cell. Of these, MCV is very important in the morphologic classification of anemias. The other red cell indices are mean cell hemoglobin (MCH), mean cell Hb concentration (MCHC), and red cell distribution width (RDW). Anemias due to a deficiency of a substance (e.g., iron) usually have a higher RDW than anemias due to a genetic defect or bone marrow disorder.
Mechanisms of anemia: Blood loss, decreased production of red blood cells, and increased destruction of red blood cells due to external factors and both hereditary and acquired internal factors (Table 12-1).
Causes of blood loss
- Acute blood loss: Often due to trauma.
- Chronic blood loss: Often due to bleeding from gastrointestinal malignancies or menstruation.
- Acute blood loss: Often due to trauma.
Causes of decreased production of red blood cells
- Disturbance of stem cells (e.g., aplastic anemia, pure red cell aplasia).
- Defective heme production (e.g., iron deficiency, thalassemias).
- Defective DNA production (e.g., vitamin B12 and folate deficiencies).
- Destruction of bone marrow (e.g., metastatic tumor).
- Disturbance of stem cells (e.g., aplastic anemia, pure red cell aplasia).
Causes of increased destruction of red blood cells due to external factors
- Antibody mediated: Etiology is either isohemagglutinin (e.g., transfusion reactions or erythroblastosis fetalis) or autoimmune.
- Trauma to red cells.
- Infectious (e.g., malaria).
- Sequestration in an enlarged spleen.
- Antibody mediated: Etiology is either isohemagglutinin (e.g., transfusion reactions or erythroblastosis fetalis) or autoimmune.
Causes of increased destruction of red blood cells due to hereditary internal factors
- Defects in cytoskeleton (e.g., hereditary spherocytosis).
- Structurally abnormal hemoglobin (e.g., sickle cell anemia, thalassemias).
- Enzyme deficiencies (e.g., glucose-6-phosphate dehydrogenase [G6PD] deficiency).
- Defects in cytoskeleton (e.g., hereditary spherocytosis).
Cause of increased destruction due to acquired defect: Paroxysmal nocturnal hemoglobinuria (PNH)
- Defect: In normal red blood cells, phosphatidylinositol glycan A (PIGA) is needed for synthesis of the glycosylphosphatidylinositol (GPI) anchor, which binds certain proteins to the cell membrane. These proteins include CD55 (also referred to as decay-accelerating factor [DAF]), and CD59, a membrane inhibitor of reactive lysis. These factors degrade complement. In patients with PNH, the loss of PIGA causes the loss of CD55 and CD59, resulting in red blood cells being more prone to lysis by complement.
- Complications of PNH: Venous thrombosis (hepatic, portal, and cerebral veins).
- Defect: In normal red blood cells, phosphatidylinositol glycan A (PIGA) is needed for synthesis of the glycosylphosphatidylinositol (GPI) anchor, which binds certain proteins to the cell membrane. These proteins include CD55 (also referred to as decay-accelerating factor [DAF]), and CD59, a membrane inhibitor of reactive lysis. These factors degrade complement. In patients with PNH, the loss of PIGA causes the loss of CD55 and CD59, resulting in red blood cells being more prone to lysis by complement.
General Mechanism of Anemia | General Cause | Specific Cause | Specific Cause |
|---|---|---|---|
Blood loss | Acute blood loss | Trauma | |
Chronic blood loss | GI malignancy, menstruation | ||
Decreased production | Defect of stem cells | Aplastic anemia, pure red cell aplasia | |
Defective heme production | Iron deficiency anemia, thalassemia | ||
Defective DNA production | Vitamin B12 and folate deficiency | ||
Destruction of bone marrow | Metastatic tumor | ||
Increased destruction | External factors: antibody mediated | Isohemagglutinin | Transfusion reactions, erythroblastosis fetalis |
Autoimmune | Warm and cold autoimmune hemolytic anemia | ||
External factors: non–antibody-mediated | Trauma | ||
Infections | |||
Sequestration | |||
Hereditary internal factors | Defect in cytoskeleton | Hereditary spherocytosis | |
Abnormal hemoglobin | Sickle cell anemia, HbC | ||
Enzyme deficiency | G6PD deficiency | ||
Acquired internal factors | PNH |
- Extravascular: Removal of red blood cells from the circulation by the phagocytic system in the spleen and liver. Complications include hemosiderin deposition in organs and jaundice and gallstones. The jaundice and gallstones result from the elevated unconjugated bilirubin associated with the hemolysis.
- Intravascular: Destruction of red blood cells within the blood vessels. Complications include unconjugated bilirubinemia, hemoglobinemia, hemoglobinuria, and hemosiderinuria. Hemoglobinemia can lead to acute tubular necrosis, which causes acute renal failure. Jaundice and gallstones can occur due to the elevated level of unconjugated bilirubin.
- Spherocytes: Present in hereditary spherocytosis and immune hemolysis.
- Schistocytes: Present in microangiopathic hemolytic anemia (e.g., thrombotic thrombocytopenic purpura [TTP], hemolytic uremic syndrome [HUS]).
- Target cells: Present in hemoglobinopathies (e.g., HbC) and obstructive liver disease.
- Teardrop cells: Present in myelofibrosis.
- Sickle cells: Present in sickle cell anemia or thalassemia minor in combination with sickle cell anemia.
- Rouleaux: Present in conditions with increased globins or decreased albumin (e.g., multiple myeloma).
- General features: Pale skin, fatigue, dyspnea on exertion, chest pain, syncope, and dizziness. Chest pain, syncope, and dizziness are all due to inadequate oxygenation of the heart and brain.
- Laboratory studies: Most important are MCV, reticulocyte count, and peripheral smear. The reticulocyte count is critical in distinguishing primary failure of red blood cell production (which has decreased reticulocytes or no reticulocytes) from increased red cell destruction (which has increased numbers of reticulocytes). Accurate assessment of the reticulocyte count requires a corrected count (reticulocyte count multiplied by the patient’s Hct/normal Hct). If the reticulocyte count is decreased, consider primary bone marrow disorders or deficiency of iron, vitamin B12, or folate. If the reticulocyte count is increased, consider hemolysis or blood loss as the source of the anemia.
Types of anemia based upon morphologic classification: There are three types of anemia based upon the patient’s MCV: microcytic, normocytic, and macrocytic.
Microcytic Anemias
Overview: MCV < 80 fL. Types include iron deficiency anemia, thalassemias, and anemia of chronic disease.
- Causes: In the United States, iron deficiency anemia is most commonly the result of chronic loss of blood due to gastrointestinal hemorrhage or menstruation. Therefore, it is often an anemia due to two mechanisms: chronic blood loss and subsequent decreased production. Other causes include dietary iron deficiency (rare in the U.S.), malabsorption of iron, or increased demands for iron (such as occur in pregnancy).
- Laboratory findings in iron deficiency anemia (Table 12-2): Decreased ferritin (storage form of iron), decreased serum iron, decreased iron in bone marrow on biopsy, increased total iron-binding capacity (TIBC), increased RDW, and decreased transferrin saturation (transferrin saturation is the ratio of serum iron to TIBC, which is normally > 20%). In iron deficiency anemia, transferrin saturation is normally > 10%.
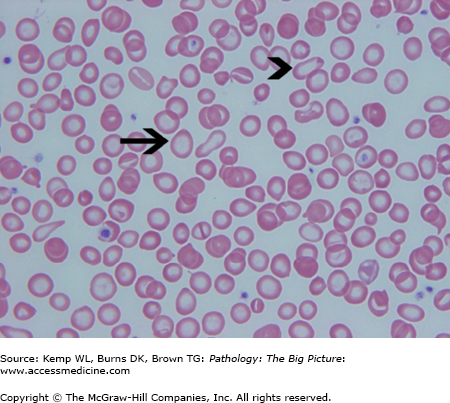
- Microscopic morphology of iron deficiency anemia: Prominent central pallor of red blood cells due to greatly reduced hemoglobin; “pencil cells” (i.e., elongated red blood cells); and marked anisocytosis of red blood cells (i.e., variation in size), which corresponds to high RDW (Figure 12-1).
Type of Microcytic Anemia | Ferritin Concentration | Serum Iron Concentration | TIBC | Transferrin Saturation |
|---|---|---|---|---|
Iron deficiency anemia | ↓ | ↓ | ↑ | ↓ |
Thalassemia | N | N | N | N |
ACD | ↑ | ↓ | ↓ | ↑ |
Basic description: Deficient production of hemoglobin due to mutation or complete loss of a globin chain. There are four copies of the α-globulin chain gene and two copies of the β-globulin chain gene; the types are α-thalassemia and β-thalassemia.
α-Thalassemia
- Basic description: Condition is due to complete loss of one or more of the four copies of the α-globulin chain gene. The loss of one copy of the gene is silent (the patient is a carrier), and the loss of two copies is asymptomatic (called α-Thal trait). The loss of three copies of the gene (called HbH disease) and of four copies (called hydrops fetalis) produces symptoms.
- Epidemiology: Prevalent in Africans and Asians.
- Important points regarding α-thalassemia
- Loss of three copies of the gene results in elevated levels of HbH (β4) and HbBart (four γ chains).
- β4 tetramers are relatively soluble and do not damage cells as do α chain tetramers.
- Loss of four α chains usually results in death in utero (hydrops fetalis).
- Loss of three copies of the gene results in elevated levels of HbH (β4) and HbBart (four γ chains).
β-Thalassemia
- Basic description: Malfunction of the gene is due to a mutation in the promoter sequence or to a mutation that causes chain termination or splicing defects, unlike in α-thalassemia, in which the disease is due to complete loss of one or more of the α-globulin chain genes. Mutations producing chain termination result in complete loss of protein (β0). Mutations in the promoter sequence may result in decreased production of the protein (β+). Mutations causing splicing defects can cause no splicing to occur (β0), or form new splice sites with some normal protein being produced (β+). Patients who are β+/β+ or β0/β0 have thalassemia major; patients who are β+/β or β0/β have thalassemia minor; and patients who are β+/β+ (minor variants, not severe enough to cause thalassemia major) or β+/β0, have thalassemia intermedia.
- Important points regarding β-thalassemia
- α-Chains are insoluble and damage red blood cells, resulting in intravascular hemolysis and ineffective erythropoiesis (apoptosis of red blood cells in marrow).
- Patients with β-thalassemia have elevated HbA2.
- α-Chains are insoluble and damage red blood cells, resulting in intravascular hemolysis and ineffective erythropoiesis (apoptosis of red blood cells in marrow).
- Epidemiology: Common in Mediterranean countries (where patients have a worse form of the disease) and in parts of Africa and Southeast Asia.
- Complications
- Thalassemia major requires blood transfusions from about the time of birth, eventually leading to secondary hemochromatosis.
- Bone marrow expansion leads to skeletal abnormalities.
- Thalassemia major requires blood transfusions from about the time of birth, eventually leading to secondary hemochromatosis.
Laboratory findings in thalassemia (see Table 12-2): Iron deficiency anemia and thalassemia are both hypochromic microcytic anemias. In thalassemia, however, the decrease in Hb is out of proportion to that of the decrease in MCV (e.g., Hb of 10.0 mg/dL and MCV of 55), whereas in iron deficiency anemia, the decrease in Hb and MCV are proportional (e.g., Hb of 10.0 mg/dL and MCV of 70).
Morphology of thalassemia
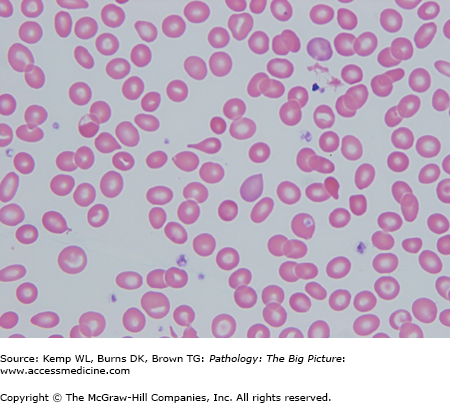
- Peripheral blood smear: Hemolysis, anisocytosis (i.e., variation in size of red blood cell), poikilocytosis (i.e., variation in shape of red blood cell), microcytosis, target cells, and reticulocytopenia (Figure 12-2).
- Other findings: “Crew-cut” skull seen on radiograph, due to bony growth; splenomegaly, hemosiderosis, or hemochromatosis.
Basic description: Anemia that occurs in the background of a chronic disease (e.g., neoplasms such as lung carcinoma or Hodgkin disease; autoimmune conditions such as rheumatoid arthritis; or chronic infections such as tuberculosis). Anemia of chronic disease is usually normocytic, but in the late stage can be microcytic.
Mechanism of anemia of chronic disease: Patients with anemia of chronic disease have elevated levels of storage iron (although serum iron is decreased) and increased ferritin and decreased TIBC, but they cannot transfer the iron from the phagocytic cells to the erythroid precursors. Elevated levels of interleukin-1 (IL-1), tumor necrosis factor (TNF), and interferon-γ associated with chronic disease processes cause decreased levels of erythropoietin and stimulate hepcidin. Hepcidin is a protein that normally impairs release of iron from storage.
Laboratory findings (see Table 12-2): Decreased TIBC, elevated ferritin, and transferrin saturation > 10%.
Other causes of microcytic anemia: Sideroblastic anemia (due to failure to synthesize iron); lead poisoning (lead blocks incorporation of iron into heme); alcohol use (ethanol inhibits enzymes in the heme synthetic pathway); and genetic diseases (e.g., Pearson disease).
Laboratory findings distinguishing microcytic anemias (see Table 12-2)
- Iron deficiency anemia: Increased RDW, TIBC, and erythrocyte protoporphyrin; decreased iron, transferrin saturation, and ferritin.
- Anemia of chronic disease: Increased ferritin and erythrocyte protoporphyrin; decreased serum iron (other tests are normal). In anemia of chronic disease, the MCV usually is > 78.
- Thalassemia minor: Elevated HbA2 helps distinguish it from thalassemia trait. Meltzer index (MCV/RBC) is > 13 in iron deficiency and < 13 in thalassemia minor.
- Sideroblastic anemia: Elevated RDW and transferrin saturation; other tests are normal.
Macrocytic Anemia
Overview: MCV > 100. A subpopulation of macrocytic anemias is referred to as megaloblastic anemia and usually has an MCV > 110. The main causes of megaloblastic anemia are deficiencies of vitamin B12 and folate.
Pathogenesis: Impaired DNA synthesis (i.e., S phase), which involves all bone marrow precursors and not just red blood cells. The impairment of DNA synthesis slows nuclear maturation but not cytoplasmic maturation, so cells have nuclear-cytoplasmic dyssynchrony.
Complications of vitamin B12 deficiency
- Anemia, thrombocytopenia, and leukopenia.
- Ineffective erythropoiesis: Patients have an adequate number of red blood cell precursors, but many cells undergo apoptosis in bone marrow because of impaired nuclear maturation.
- Neurologic deficiencies: Peripheral neuropathy and spinal cord pathology (subacute combined degeneration involving posterior and lateral columns of the spinal cord). Neurologic deficiencies are not seen in folate deficiency.
Important points
- Vitamin B12 is required for the regeneration of tetrahydrofolate. In vitamin B12 deficiency, administering folate will correct the anemia; however, it will not treat or prevent the neurologic deficiency. Administering folate bypasses the need for vitamin B12 in the production of red blood cells (i.e., there is no need to regenerate tetrahydrofolate if is constantly being supplied).
- Vitamin B12 deficiency causes elevated levels of methylmalonic acid and homocysteine.
Causes of vitamin B12 deficiency
- Pernicious anemia (see associated condition below) is the most common cause.
- Rarely dietary in the United States, but can occur in vegetarians.
- Gastrectomy (intrinsic factor required for vitamin B12 absorption is produced by parietal cells of the stomach).
- Ileal disease (e.g., Crohn disease) or resection, due to impaired resorption.
- Diphyllobothrium latum (giant fish tapeworm).
Clinical presentation of vitamin B12 deficiency
- Defect is not just limited to red blood cells, but to all cells that are growing rapidly. Therefore, symptoms of the condition are related to pancytopenia (loss of all forms of bone marrow cells) and to the loss of intestinal cells, causing diarrhea and malabsorption.
- Subacute combined degeneration is damage of the dorsal columns of the spinal cord, causing bilateral loss of proprioception, and damage of the corticospinal tracts of the spinal cord, causing spastic paresis.
Associated condition: pernicious anemia
- Basic description: Pernicious anemia is another term for autoimmune gastritis, which secondarily causes vitamin B12 deficiency because of loss of parietal cells and, thus, of intrinsic factor (IF).
- Pathogenesis of pernicious anemia: Autoimmune condition due to antibodies against parietal cells, or blocking antibodies that prevent binding of vitamin B12 to IF, or antibodies against the vitamin B12-IF complex. Any of these three antibodies prevents the absorption of vitamin B12.
Important point: The megaloblastic anemia caused by folate deficiency is clinically indistinguishable from that caused by vitamin B12 deficiency without testing of the levels of each vitamin. There are, however, some important differences between the two deficiencies.
Differences between deficiencies of folate and vitamin B12 (Table 12-3)
- Folate deficiency does not cause neurologic deficits.
- In the United States, folate deficiency is often due to dietary deficiency.
- Folate deficiency is not associated with pernicious anemia.
Feature | Vitamin B12 Deficiency | Folate Deficiency |
|---|---|---|
Form of anemia caused | Megaloblastic anemia | Megaloblastic anemia |
Cause of neurologic deficits | Yes | No |
Common cause of vitamin deficiency | Pernicious anemia | Dietary |
Microscopic morphology of megaloblastic anemia
- Peripheral blood smear: Oval macrocytes and hypersegmented neutrophils; pancytopenia (Figure 12-3).
- Bone marrow biopsy: Hyperplasia; patients have ineffective erythropoiesis in which cells are dividing, but they undergo apoptosis in the bone marrow due to impaired DNA synthesis.

Other Forms of Anemia Associated with Decreased Production
Overview: When comparing the initial outline of mechanisms of anemia to the classification of anemia based upon morphology, with the exception of aplastic anemia, infiltration of bone marrow (i.e., myelophthisic anemia), and pure red cell aplasia, most anemias due to decreased production can be categorized as microcytic or macrocytic. As microcytic and macrocytic anemias have been discussed in their own sections, this section will discuss aplastic anemia, myelophthisic anemia, and pure red cell aplasia.
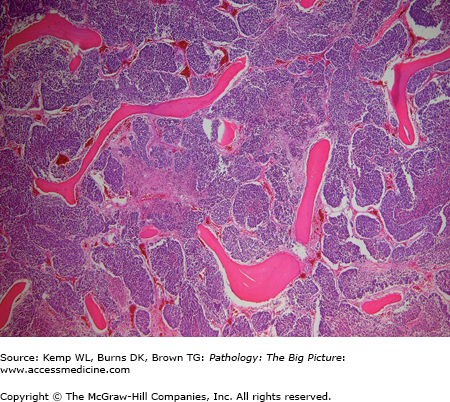
Basic description: Anemia due to absence (or near absence) of red blood cells in the bone marrow (Figure 12-4). Aplastic anemia also involves other hematopoietic cells, unless it is pure red cell aplasia, a disease associated with thymomas.

Causes of aplastic anemia
- Idiopathic (> 50% of cases).
- Secondary to drug therapy: May be a dose-dependent response (as seen with use of alkylating agents), or an idiosyncractic response (as seen with use of chloramphenicol).
- Secondary to toxins: Benzene.
- Secondary to infections: Parvovirus B19, Epstein-Barr virus (EBV), human immunodeficiency virus (HIV), and hepatitis (not hepatitis A, B, or C).
- Radiation.
- Genetic (e.g., Diamond-Blackfan syndrome).
Pathogenesis of aplastic anemia: Possibly due to suppression of stem cell function, or because the inciting agent exposes new antigens on the red blood cells, which are acted upon by the immune system.
Important point: No splenomegaly.
Laboratory findings for aplastic anemia
- Two of three findings are needed to determine a diagnosis of aplastic anemia: Neutrophil count ≤ 500/μL, platelet count < 20,000/μL, or anemia with corrected reticulocyte count of < 1%.
- Bone marrow: Cellularity < 25%.
Clinical presentation: Can be insidious. Symptoms include weakness, fatigue, dyspnea, palpitations, gingival bleeding, epistaxis, and recurrent bacterial infections.
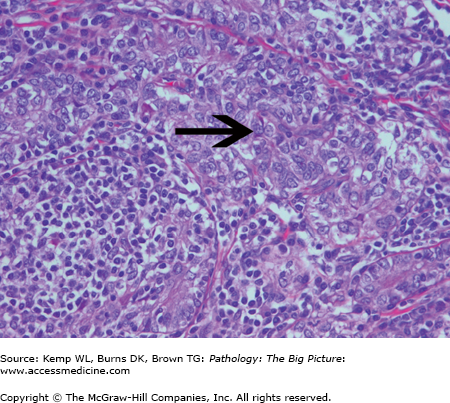
Basic description: Anemia due to space-occupying disease of the bone marrow (Figure 12-5); often associated with extramedullary hematopoiesis (i.e., hematopoiesis outside the bone marrow, often in the liver or spleen).
Figure 12-5.
Myelophthisic anemia. Myelophthisic anemia is due to a space-occupying lesion in the bone marrow, which hinders hematopoiesis. In this photomicrograph, the bone marrow space is expanded and the hematopoietic cells are obliterated by metastatic small cell lung carcinoma. Hematoxylin and eosin, 40×.
Causes of myelophthisic anemia: Metastatic tumor or idiopathic fibrosis of bone marrow.
Overview: May be primary or secondary. Secondary causes of pure red cell aplasia include thymoma (Figure 12-6), large granular lymphocytic leukemia, and autoimmune diseases.
Hemolytic Anemias
Overview: All hemolytic anemias have increased red blood cell destruction (either intravascular or extravascular), increased erythropoiesis (elevated erythropoietin and reticulocytosis), and increased iron deposition in tissues (mainly in the spleen and liver). Some patients have pigment gallstones and occasional extramedullary hematopoiesis.
Laboratory findings for hemolytic anemia: Patients with intravascular hemolysis have decreased plasma haptoglobin, increased unconjugated bilirubin, increased lactate dehydrogenase (LDH), positive urine hemosiderin, and positive urine hemoglobin. Patients with extravascular hemolysis have decreased haptoglobin and increased unconjugated bilirubin, but they do not have positive results for urine hemosiderin or urine hemoglobin.
- Congenital (e.g., enzyme deficiencies, defects of cytoskeleton, hemoglobinopathies) versus acquired (e.g., antibody-induced, mechanical fragmentation, membrane protein anchoring abnormalities, such as in PNH).
- External factors (e.g., antibody-induced, mechanical fragmentation) versus internal factors (e.g, enzyme deficiencies, hemoglobinopathies).
Important point: Hemolytic anemias can be immune-mediated or non–immune-mediated. Important laboratory testing used to distinguish these anemias are the direct and indirect Coombs tests (also called, respectively, direct antiglobulin test and indirect antiglobulin test).
- Assumption: Antibodies causing the hemolysis are present on the surface of the red blood cells.
- Mechanism: By adding antibody against Ig (which is already presumed to be on the surface of the red blood cells), cross-linking occurs, resulting in agglutination of red blood cells.
- Assumption: Antibodies causing the hemolysis are present within the plasma, but are not on the surface of the red blood cells.
- Mechanism: The indirect Coombs test requires an additional step beyond that of the direct Coombs test. Red blood cells, with an antigen the antibodies react to, are first added to the patient’s plasma. The antibodies present in the plasma bind to the added red blood cells. Then, as in the direct Coombs test, antibody is added, which cross-links the antibody bound to the red blood cells and causes agglutination of the red blood cells.
Antibody-Mediated Destruction of Red Blood Cells
Overview: Antibody-mediated destruction of red blood cells occurs through one of two main mechanisms: isohemagglutinin and autoimmune reactions. Isohemagglutinin causes of antibody-mediated destruction of red blood cells include red blood cell transfusion reactions and erythroblastosis fetalis. Red blood cell transfusion reactions occur when the recipient has an antibody against one of the donor’s red cell antigens, and can be due to ABO incompatibility or to another blood group incompatibility. Erythroblastosis fetalis occurs when there is blood group incompatibility between mother and fetus. Immunohemolytic anemias represent hemolytic anemias with an autoimmune etiology. Types of immunohemolytic anemias include warm autoimmune hemolytic anemia and cold autoimmune hemolytic anemia. This section will discuss details of ABO incompatible blood transfusion reactions, non–ABO incompatible blood transfusion reactions, erythroblastosis fetalis, warm autoimmune hemolytic anemia, cold autoimmune hemolytic anemia, and cold hemolysin hemolytic anemia.
Antibody type: IgM.
Area of hemolysis: Intravascular.
Results: Hemoglobinemia leading to acute renal failure; high mortality rate.
Important points: Anti-A and anti-B IgM are naturally occurring antibodies; therefore, an ABO incompatible red cell transfusion does not require a previous exposure to an incompatible ABO blood type.
Antibody type: Usually IgG; IgM occurs due to exposure to some non–ABO blood group antigens, but these reactions usually are not clinically significant.
Area of hemolysis: Extravascular (antibody binds to antigen on the transfused red blood cell, and the complex is removed by the spleen).
Results: Jaundice; loss of transfused unit.
Important points: Clinically significant antibodies to blood groups other than ABO (i.e., those that can cause destruction of red blood cells) usually require previous exposure to the antigen for formation. For example, the first time a patient who is D− is transfused with D+ blood may result in the formation of an antibody, and the second transfusion, in the future, will result in hemolysis. Antibodies do not always form with exposure, but some blood antigens are highly immunogenic (as is D, in which antibodies form with exposure 85% of the time).
Basic description: Condition due to maternal antibody against a fetal red blood cell antigen (usually D). When a mother who is D− has a child who is D+, the mother is exposed to the D antigen and develops anti-D antibodies. If she then has a second child who is D+, the second child can develop erythroblastosis fetalis because the maternal anti-D is IgG, which crosses the placenta and causes hemolysis of fetal red blood cells.
Presentation of erythroblastosis fetalis: Fetal hemolytic anemia, hydrops fetalis (i.e. severe fetal edema), and positive direct Coombs test. Unconjugated hyperbilirubinemia can be severe and may lead to kernicterus.
Treatment and prevention: Mothers who are D− are given RhoD (an anti-D antibody) during pregnancy. When fetal red blood cells cross into the maternal circulation, the RhoD binds to them, masking the D antigen from the mother and preventing her from forming antibodies against it.
General mechanism of warm autoimmune hemolytic anemia: IgG versus self-antigen binds red blood cells at 37°C and acts as an opsonin. Then, red blood cells are removed by the phagocytic system. The red blood cell destruction is extravascular.
Feature | Warm Autoimmune Hemolytic Anemia | Cold Autoimmune Hemolytic Anemia | |
|---|---|---|---|
Antibody type | IgG | IgM | |
Antibody versus | Self antigens | I and i | |
Coombs test | Positive | Negative | |
Causes | Idiopathic | Acute | Mycoplasma, EBV |
B-cell neoplasms | Chronic | B-cell neoplasms | |
SLE | |||
Drug reactions | |||
Mechanisms of drug-induced hemolysis (a form of warm autoimmune hemolytic anemia): There are three mechanisms of warm autoimmune hemolytic anemia due to drug-induced hemolysis.
- Stimulation of autoantibody production (e.g., due to α-methyldopa).
- Drugs act as a hapten and induce antibody formation (e.g., due to penicillin).
- Immune complex deposition: Antibody binds to the drug, and complex deposits on surface of the red blood cell.
Causes of production of antibody versus self-antigens on red blood cells
- Primary: 60% of cases of warm autoimmune hemolytic anemia are idiopathic, with no identifiable cause.
- Secondary: Due to B-cell neoplasms, systemic lupus erythematosus, and drugs.
Microscopic morphology of warm autoimmune hemolytic anemia: Spherocytes in peripheral blood smear.
Clinical presentation of warm autoimmune hemolytic anemia
- Anemia, jaundice, elevated reticulocyte count. Patient may have splenomegaly.
- Coombs test is positive.
Mechanism: IgM binds to red blood cells. IgM binding to I antigen is associated with Mycoplasma infections. IgM binding to i antigen is associated with EBV infections. The IgM binds at 30°C and also binds complement. The IgM releases at 37°C, leaving C3b, which acts as opsonin.
Causes of cold autoimmune hemolytic anemia
- Acute form: Mycoplasma pneumoniae; infectious mononucleosis (EBV). Acute cold autoimmune hemolytic anemia is usually self-limited.
- Chronic form: Idiopathic; B-cell neoplasms.
Morphology: Spherocytes.
Laboratory finding: Coombs test is negative.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


