Hematologic Disorders
INTRODUCTION
Blood, one of the body’s major fluid tissues, continuously circulates through the heart and blood vessels, carrying vital elements to every part of the body.
BLOOD BASICS
Blood performs several vital functions through its special components: the liquid portion (plasma) and the formed constituents (erythrocytes, leukocytes, and thrombocytes) that are suspended in it. Erythrocytes (red blood cells [RBCs]) carry oxygen to the tissues and remove carbon dioxide from them. Leukocytes (white blood cells [WBCs]) act in inflammatory and immune responses. Plasma (a clear, straw-colored fluid) carries antibodies and nutrients to tissues and carries waste away; plasma coagulation factors and thrombocytes (platelets) control clotting. (See Coagulation factors, page 456.)
Typically, the average person has 5 to 6 L of circulating blood, which constitute 5% to 7% of body weight (as much as 10% in premature neonates). Blood is three to five times more viscous than water, with an alkaline pH of 7.35 to 7.45, and is either bright red (arterial blood) or dark red (venous blood), depending on the degree of oxygen saturation and the hemoglobin (Hb) level.
FORMATION AND CHARACTERISTICS
Hematopoiesis, the process of blood formation, occurs primarily in the bone marrow of the femur, sternum, and vertebrae, where primitive blood cells (stem cells) produce the precursors of erythrocytes (normoblasts), leukocytes, and thrombocytes. During embryonic development, blood cells are derived from mesenchyma and form in the yolk sac. As the fetus matures, blood cells are produced in the liver, the spleen, and the thymus; by the fifth month of gestation, blood cells also begin to form in bone marrow. After birth, blood cells are usually produced only in the marrow.
BLOOD’S FUNCTION
The most important function of blood is to transport oxygen (bound to RBCs inside Hb) from the lungs to the body tissues and to return carbon dioxide from these tissues to the lungs. Blood also performs the following functions:
production and delivery of antibodies (by WBCs) formed by plasma cells and lymphocytes
transportation of granulocytes and monocytes to defend the body against pathogens by phagocytosis
immunity against viruses and cancer cells through sensitized lymphocytes
provision of complement, a group of immunologically important protein substances in plasma essential for immune and inflammatory responses
Blood’s other functions include control of hemostasis by platelets, plasma, and coagulation factors that repair tissue injuries and prevent or halt bleeding; acid-base and fluid balance; regulation of body temperature by carrying off excess heat generated by the internal organs for dissipation through the skin; and transportation of nutrients and regulatory hormones to body tissues and of metabolic wastes to the organs of excretion (kidneys, lungs, and skin).
BLOOD DYSFUNCTION
Because of the rapid reproduction of bone marrow cells and the short life span and minimal storage in the bone marrow of circulating cells, bone marrow cells and their precursors are particularly vulnerable to physiologic changes that can affect cell production, or hematopoiesis. Resulting blood disorders may be primary or secondary, quantitative or qualitative, or both; they may involve some or all blood components. Quantitative blood disorders result from increased or decreased cell production or cell destruction; qualitative blood disorders stem from intrinsic cell abnormalities or plasma component dysfunction. Specific causes of blood disorders include trauma, chronic disease, surgery, malnutrition, drugs, exposure to toxins and radiation, and genetic and congenital defects that disrupt production and function. For example, depressed bone marrow production or mechanical destruction of mature blood cells can reduce the number of RBCs, platelets, and granulocytes, resulting in pancytopenia (anemia, thrombocytopenia, and granulocytopenia). Increased production of multiple bone marrow components can follow myeloproliferative disorders.
ERYTHROPOIESIS
The tissues’ demand for oxygen and the blood cells’ ability to deliver it regulate RBC production, or erythropoiesis. Consequently, hypoxia (or tissue anoxia) stimulates RBC production by triggering the formation and release of erythropoietin, a hormone (produced by the kidneys) that activates bone marrow to produce RBCs. Erythropoiesis may also be stimulated by androgens (which accounts for the higher RBC counts in men). RBCs have a life span of approximately 120 days.
Coagulation factors | ||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
The actual formation of an erythrocyte begins with an uncommitted stem cell that may eventually develop into an RBC or a WBC. Such formation requires certain vitamins—B12 and folic acid—and minerals, such as copper, cobalt, and especially iron, which is vital to hemoglobin’s oxygen-carrying capacity. Iron is obtained from various foods and is absorbed in the duodenum and upper jejunum, leaving the excess for temporary storage in reticuloendothelial cells, especially those in the liver. Iron excess is stored as ferritin and hemosiderin until it’s released for use in the bone marrow to form new RBCs.
In older adults, fatty bone marrow replaces some active cell-forming marrow—first in the long bones and later in the flat bones. The altered bone marrow can’t increase erythrocyte production as readily as before in response to such stimuli as hormones, anoxia, hemorrhage, and hemolysis.
RBC DISORDERS
RBC disorders include quantitative and qualitative abnormalities. Deficiency of RBCs (anemia) can follow any condition that destroys or inhibits the formation of these cells. Common factors leading to this deficiency include:
chronic illnesses, such as renal disease, cancer, and chronic infections
congenital or acquired defects that cause bone marrow aplasia and suppress general hematopoiesis (aplastic anemia) or erythropoiesis
deficiencies of vitamins (vitamin B12 deficiency or pernicious anemia) or minerals (iron, folic acid, copper, and cobalt deficiency anemias) that cause inadequate RBC production
drugs, toxins, and ionizing radiation
excessive chronic or acute blood loss (posthemorrhagic anemia)
intrinsically or extrinsically defective RBCs (sickle cell anemia and hemolytic transfusion reaction)
metabolic abnormalities (sideroblastic anemia)
Comparatively few conditions lead to excessive numbers of RBCs:
abnormal proliferation of all bone marrow elements (polycythemia vera), especially RBC mass
a single-element abnormality (for instance, an increase in RBCs that results from erythropoietin excess, which in turn results from hypoxemia, hypertension, or pulmonary disease)
decreased plasma cell volume, which produces a corresponding relative increase in RBC concentration (such as through the use of drugs)
FUNCTION OF WBCS
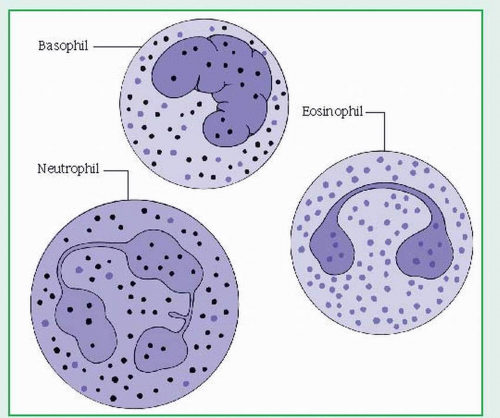
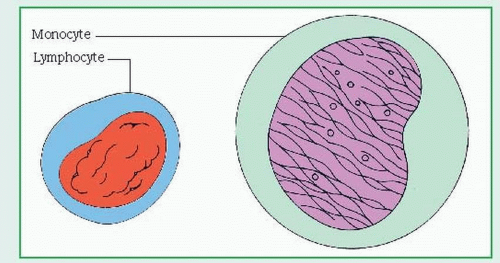
WBCs, or leukocytes, protect the body against harmful bacteria and infection and are classified as granular leukocytes (basophils, neutrophils, and eosinophils) or nongranular leukocytes (lymphocytes, monocytes, and plasma cells). (See Two types of leukocytes.) Usually, WBCs are produced in bone marrow; lymphocytes and plasma cells are produced in lymphoid tissue as well. Neutrophils have a circulating half-life of less than 6 hours; some lymphocytes may survive for weeks or months. Normally, WBCs number between 5,000 and 10,000/µl.
There are six types of WBCs:
Neutrophils, the predominant form of granulocyte, make up about 60% of WBCs; they help devour invading organisms by phagocytosis.
Eosinophils, minor granulocytes, may defend against parasites and lung and skin infections and act in allergic reactions. They account for 1% to 5% of the total WBC count.
Basophils, minor granulocytes, may release heparin and histamine into the blood and participate in delayed hypersensitivity reactions. They account for 0% to 1% of the total WBC count.
Monocytes, along with neutrophils, help devour invading organisms by phagocytosis. They help process antigens for lymphocytes and form macrophages in the tissues; they account for 1% to 6% of the total WBC count.
Lymphocytes occur as B cells and T cells. B cells aid antibody synthesis; T cells regulate cell-mediated immunity. They account for 20% to 40% of the total WBC count.
Plasma cells develop from lymphocytes, reside in the tissue, and produce antibodies. Plasma cells do not normally circulate in the blood.
Two types of leukocytes
Leukocytes vary in size, shape, and number.
Granular leukocytes
Granular leukocytes (granulocytes) are the most numerous and include basophils, containing cytoplasmic granules that stain readily with alkaline dyes; eosinophils, which stain with acidic dyes; and neutrophils, which are finely granular and recognizable by their multinucleated appearance.
Nongranular leukocytes
Lymphocytes and monocytes have few, if any, granulated particles in the cytoplasm.
 GRANULAR LEUKOCYTES |
 NONGRANULAR LEUKOCYTES |
A temporary increase in production and release of mature WBCs (leukemic reaction) is a normal response to infection. However, an excessive number of immature WBC precursors and their accumulation in bone marrow or lymphoid tissue is characteristic of leukemia. These nonfunctioning WBCs (blasts) provide no protection against infection; crowd out RBCs, platelets, and mature WBCs; and spill into the bloodstream, sometimes infiltrating organs and impairing function.
WBC deficiencies may reflect inadequate cell production, drug reactions, ionizing radiation, infiltrated bone marrow (cancer), congenital defects, aplastic anemia, folic acid deficiency, or hypersplenism. The major WBC deficiencies are granulocytopenia, lymphocytopenia, and monocytopenia.
PLATELETS, PLASMA, AND CLOTTING
Platelets are small (2 to 4 microns in diameter), colorless, disk-shaped cytoplasmic fragments split from cells in bone marrow called megakaryocytes. The normal platelet concentration is 150,000 to 400,000/µl. These fragments, which have a life span of approximately 10 days, perform three vital functions:
initiate vasoconstriction of damaged blood vessels to minimize blood loss
form hemostatic plugs in injured blood vessels
with plasma, provide materials that accelerate blood coagulation—notably platelet factor 3
Plasma consists mainly of proteins (chiefly albumin, globulin, and fibrinogen) held in aqueous suspension. Other components of plasma include glucose, lipids, amino acids, electrolytes, pigments, hormones, respiratory gases (oxygen and carbon dioxide), and products of metabolism, such as urea, uric acid, creatinine, and lactic acid. Its fluid characteristics— including osmotic pressure, viscosity, and suspension qualities—depend on its protein content. Plasma components regulate acid-base balance and immune responses and mediate coagulation and nutrition.
In a complex process called hemostasis, platelets, plasma, and coagulation factors interact to control bleeding.
HEMOSTASIS AND THE CLOTTING MECHANISM
Hemostasis is the complex process by which the body controls bleeding. When a blood vessel ruptures, local vasoconstriction and platelet clumping (aggregation) at the injury site initially help prevent hemorrhage. This activation of the coagulation system, called extrinsic cascade, requires release of tissue thromboplastin from the damaged cells. However, formation of a more stable clot requires initiation of the complex clotting mechanism known as the intrinsic cascade system. When endothelial vessel injury or a foreign body in the bloodstream activates this system, activating factor XII triggers clotting. In the final common pathway, prothrombin is converted to thrombin. Thrombin acts on fibrinogen to form fibrin, the basis of a clot.
THERAPY WITH BLOOD COMPONENTS
Because of improved methods of collection, component separation, and storage, blood transfusions are being used more effectively than ever. Separating blood into components permits a single unit of blood to benefit several patients with different hematologic abnormalities. Component therapy allows replacement of a specific blood component without risking reactions from other components.
Blood typing, crossmatching, and human leukocyte antigen (HLA) typing are compatibility tests used to ensure safe, effective replacement therapy and minimize the risk of transfusion reactions. Blood typing determines the antigens present in the patient’s RBCs by reaction with standardized sera. (Critical antigen groups are those of ABO and Rh factor.) Crossmatching the patient’s blood with transfusion blood provides some assurance that the patient doesn’t have antibodies against donor red cells. HLA typing may be helpful for the patient who needs long-term transfusion therapy or frequent platelet transfusions, but usually, only family members can provide an appropriate match because antigenic properties are genetically determined.
BONE MARROW TRANSPLANTATION
Bone marrow transplantation is used to treat acute leukemia, aplastic anemia, severe combined immunodeficiency disease, Nezelof syndrome, and Wiskott-Aldrich syndrome. In this procedure, marrow from a twin or another HLAidentical donor (usually a sibling) is transfused in an attempt to repopulate the recipient’s bone marrow with normal cells.
A hematologic disorder can affect nearly every aspect of the patient’s life, perhaps resulting in life-threatening emergencies that require prompt medical treatment. This is particularly true of patients with such diseases as hemophilia and thalassemia major, diseases for which no cure is available. Astute, sensitive care founded on a firm understanding of hematologic basics can help the patient survive such illnesses. In situations with poor prognoses, the patient may need to make many adjustments to maintain an optimal quality of life.
ANEMIAS
Pernicious anemia
Pernicious anemia, also known as vitamin B,12 deficiency, is a type of megaloblastic anemia characterized by decreased gastric production
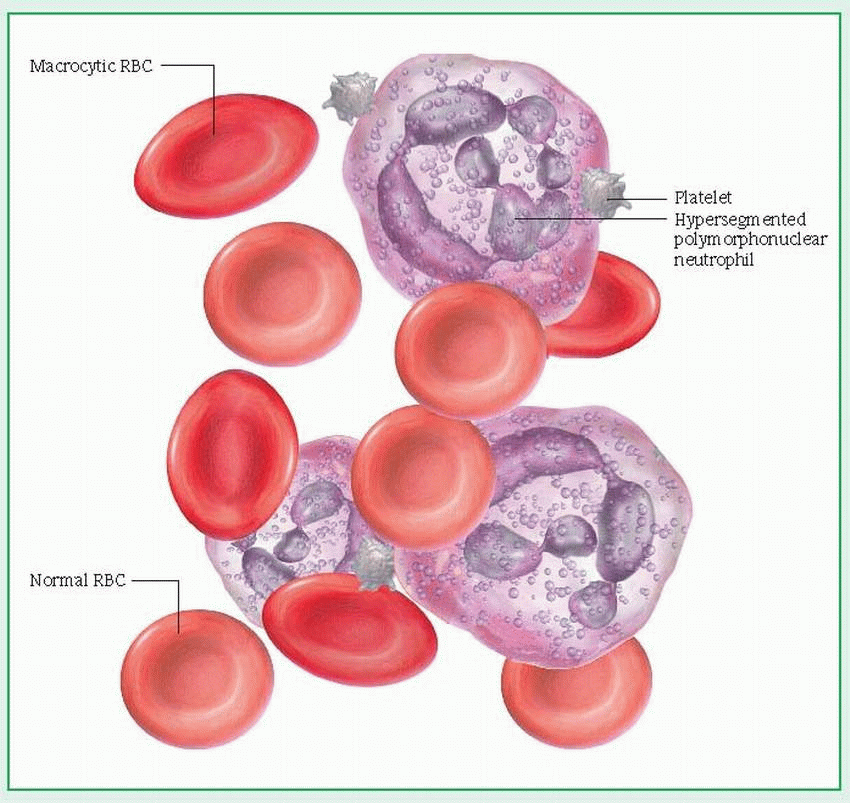
of hydrochloric acid and deficiency of intrinsic factor (IF), a substance normally secreted by the parietal cells of the gastric mucosa that’s essential for vitamin B12 absorption in the ileum. The resulting deficiency of vitamin B12 causes serious neurologic, gastric, and intestinal abnormalities. Untreated pernicious anemia may lead to permanent neurologic disability and death. (See Peripheral blood smear in pernicious anemia.)
of hydrochloric acid and deficiency of intrinsic factor (IF), a substance normally secreted by the parietal cells of the gastric mucosa that’s essential for vitamin B12 absorption in the ileum. The resulting deficiency of vitamin B12 causes serious neurologic, gastric, and intestinal abnormalities. Untreated pernicious anemia may lead to permanent neurologic disability and death. (See Peripheral blood smear in pernicious anemia.)
CAUSES AND INCIDENCE
Familial incidence of pernicious anemia suggests a genetic predisposition. (It may involve an inherited single dominant autosomal factor.) Significantly higher incidence in patients with immunologically related endocrine diseases, such as thyroiditis, myxedema, and Graves’ disease, seems to support a widely held theory that an inherited autoimmune response causes gastric mucosal atrophy and, therefore, deficiency of hydrochloric acid and IF. IF deficiency impairs vitamin B12 absorption. The resultant vitamin B12 deficiency inhibits cell growth, particularly of red blood cells (RBCs), leading to insufficient and deformed RBCs with poor oxygen-carrying capacity. It also impairs myelin formation, causing neurologic damage. Iatrogenic induction can follow partial gastrectomy.
Juvenile pernicious anemia, occurring in children younger than age 10, stems from a congenital stomach disorder that causes secretion of abnormal IF.
With age, vitamin B,12 absorption may also diminish, resulting in reduced erythrocyte mass and decreased hemoglobin (Hb) levels and hematocrit (HCT).
Pernicious anemia primarily affects people of northern European ancestry. It’s rare in children and infants. Onset typically occurs after age 35, and incidence increases with age. It affects about 2% of people older than age 60.
COMPLICATIONS
Paralysis
Psychotic behavior
Loss of bowel and bladder sphincter control
SIGNS AND SYMPTOMS
Characteristically, pernicious anemia has an insidious onset but eventually causes an unmistakable triad of symptoms: weakness, sore tongue, and numbness and tingling in the extremities. The lips, gums, and tongue appear markedly bloodless. Hemolysis-induced hyperbilirubinemia may cause faintly jaundiced sclera and pale to bright yellow skin. In addition, the patient may become highly susceptible to infection, especially of the genitourinary tract.
Other systemic symptoms of pernicious anemia include the following:
GI—Gastric mucosal atrophy and decreased hydrochloric acid production disturb digestion and lead to nausea, vomiting, anorexia, weight loss, flatulence, diarrhea, and constipation. Gingival bleeding and tongue inflammation may hinder eating and intensify anorexia.
Central nervous system (CNS)—Demyelination caused by vitamin B12 deficiency initially affects the peripheral nerves but gradually extends to the spinal cord. Consequently, the neurologic effects of pernicious anemia may include neuritis; weakness in extremities; peripheral numbness and paresthesia; disturbed position sense; lack of coordination; ataxia; impaired fine finger movement; positive Babinski’s and Romberg’s signs; light-headedness; altered vision (diplopia and blurred vision), taste, and hearing (tinnitus); optic muscle atrophy; loss of bowel and bladder control; and, in males, impotence. Its effects on the nervous system may also produce irritability, poor memory, headache, depression, and delirium. Although some of these symptoms are temporary, irreversible CNS changes may have occurred before treatment.
Cardiovascular—Increasingly fragile cell membranes induce widespread destruction of RBCs, resulting in low Hb levels. The impaired oxygen-carrying capacity of the blood secondary to lowered Hb leads to weakness, fatigue, and light-headedness. Compensatory increased cardiac output results in palpitations, wide pulse pressure, dyspnea, orthopnea, tachycardia, premature beats and, eventually, heart failure.
Musculoskeletal—Scissors gait can also occur as a late sign of untreated anemia.
DIAGNOSIS
A positive family history, typical ethnic heritage, and results of blood studies, bone marrow aspiration, gastric analysis, and the Schilling test establish the diagnosis. (See Tests for blood composition, production, and function.) Laboratory screening must rule out other anemias with similar symptoms, such as folic acid deficiency anemia, because treatment differs. Diagnosis must also rule out vitamin B12 deficiency resulting from malabsorption due to GI disorders, gastric surgery, radiation, or drug therapy.
Blood study results that suggest pernicious anemia include:
▪decreased Hb levels (4 to 5 g/dl) and de creased RBC count
Hb levels drop 1 to 2 g/dl in elderly men, and HCT may decrease slightly in both men and women. These changes reflect decreased bone marrow and hematopoiesis and (in men) decreased androgen levels; they aren’t an indicator of pernicious anemia.
increased mean corpuscular volume (greater than 120/µl); because larger-than-normal RBCs each contain increased amounts of Hb, mean corpuscular Hb concentration is also increased
possible low white blood cell and platelet counts and large, malformed platelets
serum vitamin B12 assay levels less than 0.1 mcg/ml
elevated serum lactate dehydrogenase levels.
Bone marrow aspiration reveals erythroid hyperplasia (crowded red bone marrow), with increased numbers of megaloblasts but few normally developing RBCs. Gastric analysis shows absence of free hydrochloric acid after histamine or pentagastrin injection.
The Schilling test was once the definitive diagnostic test for pernicious anemia. It is rarely used now because it involves the injection of radioactive vitamin B12. Now, vitamin B,2 levels are measured along with tests for the presence of IF and antiparietal cell antibodies.
In addition, a complete blood count with decreased Hb level and RBC count will reveal anemia. Elevated homocysteine and methylmalonic acid (MMA) levels may indicate pernicious anemia. In some cases, bone marrow aspiration may be performed. If pernicious anemia is present the aspirate will show erythroid hyperplasia, increased numbers of megaloblasts but few normally developing RBCs.
Tests for blood composition, production, and function
Overall composition
Peripheral blood smear shows maturity and morphologic characteristics of blood elements and determines qualitative abnormalities.
Complete blood count determines the actual number of blood elements in relation to volume and quantifies abnormalities.
Bone marrow aspiration or biopsy allows evaluation of hematopoiesis by showing blood elements and precursors, and abnormal or malignant cells.
Red blood cell function
Hematocrit, or packed cell volume, measures the percentage of red blood cells (RBCs) per fluid volume of whole blood.
Hemoglobin (Hb) measures the amount (grams) of Hb per deciliter of blood, to determine oxygen-carrying capacity.
Reticulocyte count assesses RBC production by determining concentration of this erythrocyte precursor.
Schilling test determines absorption of vitamin B12 (necessary for erythropoiesis) by measuring excretion of radioactive B12 in the urine.
Mean corpuscular volume describes the RBC in terms of size.
Mean corpuscular Hb determines the average amount of Hb per RBC.
Mean corpuscular Hb concentration establishes the average Hb concentration in 1 dl of packed RBCs.
Sucrose hemolysis test assesses the susceptibility of RBCs to hemolyze with complement.
Direct Coombs’ test demonstrates the presence of immunoglobulin G (IgG) antibodies (such as antibodies to Rh factor) or, possibly, complement on circulating RBCs.
Indirect Coombs’ test, a two-step test, detects the presence of IgG antibodies in the serum.
Sideroblast test detects stainable iron (available for Hb synthesis) in normoblastic RBCs.
Hb electrophoresis demonstrates abnormal Hb such as sickle cell anemia.
Hemostasis
Platelet count determines the number of platelets.
Bleeding time (Ivy bleeding time) assesses the capacity for platelets to stop bleeding in capillaries and small vessels.
Prothrombin time (Quick’s test, pro time) assists in evaluation of thrombin generation (extrinsic clotting mechanism).
Partial thromboplastin time aids evaluation of the adequacy of plasma-clotting factors (intrinsic clotting mechanism).
International Normalized Ratio (INR) normalizes ratios between labs.
Fibrin degradation products (fibrin split products, FDPs) test the amount of clot breakdown products in serum.
Thrombin time detects abnormalities in thrombin fibrinogen reaction.
Fibrinogen (factor I) measures this coagulation factor in plasma.
D-dimer test determines if FDPs are from normal mechanisms or excessive fibrinolysis and is commonly used to diagnose disseminated intravascular coagulation.
White blood cell function
White blood cell (WBC) count and differential establishes quantity and matu rity of WBC elements (neutrophils [called polymorphonuclear granulocytes or bands], basophils, eosinophils, lymphocytes, monocytes).
Quantified CD4+:CD8+ T lymphocytes determines helper:suppressor ratio, which is important to immune function and is decreased in HIV infection.
Plasma
Erythrocyte sedimentation rate measures the rate of RBCs settling from plasma and may reflect infection.
Electrophoresis of serum proteins determines the amount of various serum proteins (classified by mobility in response to an electrical field). It’s commonly used to diagnose plasma cell myeloma.
Immunoelectrophoresis of serum proteins separates and classifies serum antibodies (immunoglobulins) through specific antisera.
TREATMENT
Early parenteral vitamin B12 replacement can reverse pernicious anemia, minimize complications and, possibly, prevent permanent neurologic damage. An initial high dose of parenteral vitamin B12 causes rapid RBC regeneration. Within 2 weeks, Hb levels should rise to normal, and the patient’s condition should markedly improve. Because rapid cell regeneration increases the patient’s iron and folate requirements, concomitant iron and folic acid replacement is necessary to prevent iron deficiency anemia.
After the patient’s condition improves, the vitamin B12 dosage can be decreased to maintenance levels and given monthly. Because such injections must be continued for life, the patient should learn self-administration of vitamin B12.
A new vitamin B12 nasal gel that is applied once a week may be used in place of injections to maintain vitamin B12 levels.
If anemia causes extreme fatigue, the patient may require bed rest until Hb levels rise. If Hb levels are dangerously low, he may need blood transfusions. Digoxin, a diuretic, and a lowsodium diet may be necessary for a patient with heart failure. Most important is the replacement of vitamin B12 to control the condition that led to this failure. Antibiotics help combat accompanying infections.
SPECIAL CONSIDERATIONS
Supportive measures minimize the risk of complications and speed recovery. Patient and family teaching can promote compliance with lifelong vitamin B12 replacement.
If the patient has severe anemia, plan activities, rest periods, and necessary diagnostic tests to conserve his energy. Monitor pulse rate often; tachycardia means his activities are too strenuous.
To ensure accurate Schilling test results, make sure that all urine over a 24-hour period is collected and that the specimens are uncontaminated.
Warn the patient to guard against infections and tell him to report signs of infection promptly, especially pulmonary and urinary tract infections, because the patient’s weakened condition may increase susceptibility.
Provide a well-balanced diet, including foods high in vitamin B12 (meat, liver, fish, eggs, and milk). Offer between-meal snacks and encourage the family to bring favorite foods from home.
Because a sore mouth and tongue make eating painful, ask the dietitian to avoid giving the patient irritating foods. If these symptoms make talking difficult, supply a pad and pencil or some other aid to facilitate nonverbal communication; explain this problem to the family. Provide diluted mouthwash or, with severe conditions, swab the patient’s mouth with tap water or warm saline solution or use topical anesthetic mouthwash.
Warn the patient with a sensory deficit not to use a heating pad, because it may cause burns.
If the patient is incontinent, establish a regular bowel and bladder routine. After the patient is discharged, a home health care nurse should follow up on this schedule and make adjustments, as needed.
If neurologic damage causes behavioral problems, assess mental and neurologic status often; if needed, give tranquilizers, as ordered, and apply wrist or jacket restraint and utilize bed alert alarm at night.
Stress that vitamin B12 replacement isn’t a permanent cure and that these injections must be continued for life, even after symptoms subside.
To prevent vitamin B12 deficiency, emphasize the importance of vitamin B12 supplements for patients who have had extensive gastric resections or who follow strict vegetarian or vegan diets.
Folic acid deficiency anemia
Folic acid is a water-soluble vitamin that is rapidly excreted; body stores are limited to a few weeks. It is therefore relatively easy to develop a folic acid deficiency.
Folic acid deficiency anemia is a common, slowly progressive, megaloblastic anemia. It usually occurs in infants, adolescents, pregnant and lactating females, alcoholics, elderly people, and people with malignant or intestinal diseases.
CAUSES AND INCIDENCE
Folic acid deficiency anemia may result from:
alcohol abuse (alcohol may suppress metabolic effects of folate)
poor diet (common in alcoholics, elderly people living alone, and infants, especially those with infections or diarrhea)
impaired absorption (due to intestinal dysfunction from disorders such as celiac disease, tropical sprue, regional jejunitis, or bowel resection)
bacteria competing for available folic acid
excessive cooking, which can destroy a high percentage of folic acids in foods (See Preventing folic acid deficiency anemia.)
limited storage capacity in infants
prolonged drug therapy (anticonvulsants and estrogens)
increased folic acid requirements during pregnancy; during rapid growth in infancy (common because of recent increase in survival of premature infants); during childhood and adolescence (because of general use of folatepoor cow’s milk); and in patients with neoplastic diseases and some skin diseases (chronic exfoliative dermatitis)
It’s estimated that 10% of the United States population has low folate stores.
SIGNS AND SYMPTOMS
Folic acid deficiency anemia gradually produces clinical features characteristic of other megaloblastic anemias, without the neurologic manifestations: progressive fatigue, shortness of breath, palpitations, weakness, glossitis, mouth ulcers, nausea, anorexia, headache, fainting, irritability, forgetfulness, pallor, and slight jaundice. Folic acid deficiency anemia doesn’t cause neurologic impairment unless it’s associated with vitamin B12 deficiency, as in pernicious anemia.
DIAGNOSIS
A complete blood count (CBC) with significant findings including macrocytosis, decreased reticulocyte count, abnormal platelets, low hemoglobin level, normal or high vitamin B12 level, and serum folate less than 3 ng/ml confirm the diagnosis.
TREATMENT
Treatment consists primarily of folic acid supplements and elimination of contributing causes. Folic acid supplements may be given orally or parenterally (to patients who are severely ill, have malabsorption, or are unable to take oral medication). Many patients respond favorably to a well-balanced diet. Vitamin B12 deficiency should be ruled out before administering folic acid. If the patient has combined B12 and folate deficiencies, folic acid replenishment alone may aggravate neurologic dysfunction.
SPECIAL CONSIDERATIONS
Teach the patient to meet daily folic acid requirements by including a food from each food group in every meal. If he has a severe deficiency, explain that diet only reinforces fo lic acid supplementation and isn’t therapeutic by itself. Urge compliance with the prescribed course of therapy. Advise him not to stop taking the supplements when he begins to feel better.
If the patient has glossitis, emphasize the importance of good oral hygiene. Suggest regular use of mild or diluted mouthwash and a soft toothbrush.
Watch fluid and electrolyte balance, particularly in the patient who has severe diarrhea and is receiving parenteral fluid replacement therapy.
Because anemia causes severe fatigue, schedule regular rest periods until the patient is able to resume normal activity.
Folic acid (pteroylglutamic acid, folacin) is found in most body tissues, where it acts as a coenzyme in metabolic processes involving one carbon transfer. It’s essential for formation and maturation of red blood cells and for synthesis of deoxyribonucleic acid. Although its body stores are relatively small (about 70 mg), this vitamin is plentiful in most well-balanced diets.
However, because folic acid is watersoluble and heat-labile, it’s easily destroyed by cooking. Also, approximately 20% of folic acid intake is excreted unabsorbed. Insufficient daily folic acid intake (less than 50 mcg/day) usually induces folic acid deficiency within 4 months. To prevent folic acid deficiency anemia, foods high in folic acid content should be chosen, such as those listed below:
Food | mcg/100 g |
Asparagus spears | 109 |
Beef liver | 294 |
Broccoli spears | 54 |
Collards (cooked) | 102 |
Mushrooms | 24 |
Oatmeal | 33 |
Peanut butter | 57 |
Red beans | 180 |
Wheat germ | 305 |
Emphasize the importance of a well-balanced diet high in folic acid. Identify alcoholics with poor dietary habits and try to arrange for appropriate counseling. Tell mothers who aren’t breast-feeding to use commercially prepared formulas.
Aplastic anemias
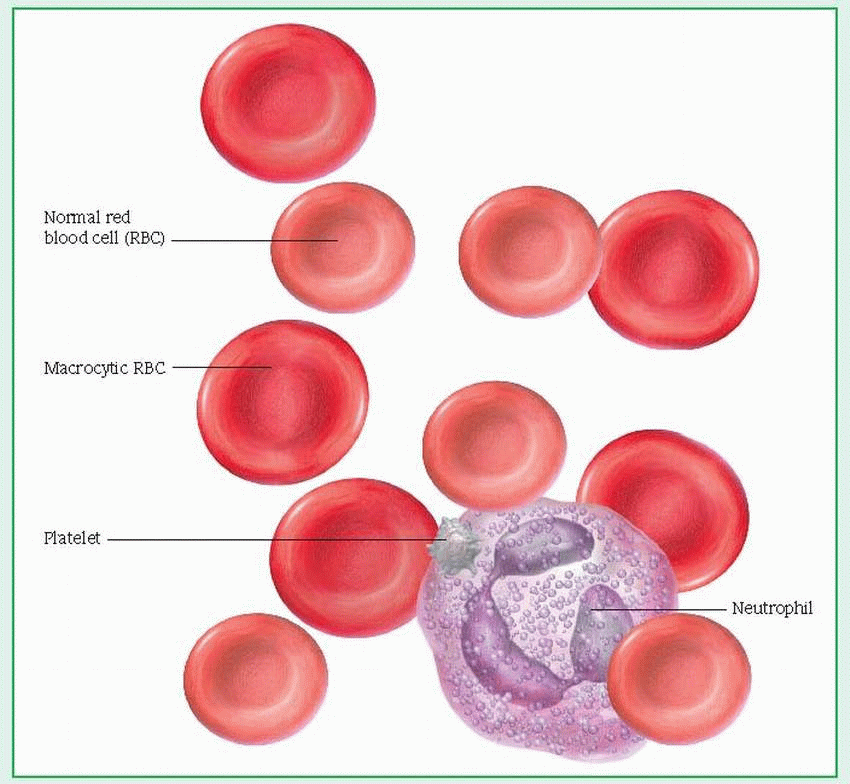
Aplastic, or hypoplastic, anemias result from injury to or destruction of stem cells in bone marrow or the bone marrow matrix, causing pancytopenia (anemia, granulocytopenia, and thrombocytopenia) and bone marrow hypoplasia. (See Peripheral blood smear in aplastic anemia.) Although commonly used interchangeably with other terms for bone marrow failure, aplastic anemia properly refers to pancytopenia resulting from the decreased functional capacity of a hypoplastic, fatty bone marrow. These disorders generally produce fatal bleeding or infection, particularly when they’re idiopathic or stem from chloramphenicol or from infectious hepatitis. Mortality for aplastic anemias with severe pancytopenia is 80% to 90%.
CAUSES AND INCIDENCE
Aplastic anemias usually develop when damaged or destroyed stem cells (which develop into red blood cells [RBCs], white blood cells, and platelets) inhibit RBC production. Less commonly, they develop when damaged bone marrow microvasculature creates an unfavorable environment for cell growth and maturation. About one-half of such anemias result from drugs (antibiotics, anticonvulsants, anti-inflammatory drugs, antineoplastics, diuretics, phenothiazines, antidiabetics, and antithyroid drugs), toxic agents (such as benzene and chloramphenicol), or radiation. The rest may result from immunologic factors (unconfirmed), severe disease (especially hepatitis), viral infection (especially in
children), or preleukemic and neoplastic infiltration of bone marrow.
children), or preleukemic and neoplastic infiltration of bone marrow.
Idiopathic anemias may be congenital. Two such forms of aplastic anemia have been identified: Congenital hypoplastic anemia (Blackfan-Diamond anemia) develops between ages 2 and 3 months; Fanconi’s syndrome, between birth and age 10. In Fanconi’s syndrome, chromosomal abnormalities are usually associated with multiple congenital anomalies, such as dwarfism, and hypoplasia of the kidneys and spleen. In the absence of a consistent familial or genetic history of aplastic anemia, researchers suspect that these congenital abnormalities result from an induced change in the fetus’ development.
Incidence is 0.6 to 6.1 cases per 1 million people in the United States. There is no racial predilection.
COMPLICATIONS
Life-threatening hemorrhage
Secondary opportunistic infections
SIGNS AND SYMPTOMS
Clinical features of aplastic anemias vary with the severity of pancytopenia but develop insidiously in many cases. Anemic symptoms include progressive weakness and fatigue, shortness of breath, headache, pallor and, ultimately, tachycardia and heart failure. Thrombocytopenia leads to ecchymosis, petechiae, and hemorrhage, especially from the mucous membranes (nose, gums, rectum, and vagina) or into the retina or central nervous system. Neutropenia may lead to infection (fever, oral and rectal ulcers, and sore throat) but without characteristic inflammation.
DIAGNOSIS
Confirmation of aplastic anemia requires a series of laboratory tests:
RBCs are usually normochromic and normocytic (although macrocytosis [larger-than-normal erythrocytes] and anisocytosis [excessive variation in erythrocyte size] may exist), with a total count of 1 million/µl or less. Absolute reticulocyte count is very low.
Serum iron level is elevated (unless bleeding occurs), but total iron-binding capacity is normal or slightly reduced. Hemosiderin (a derivative of hemoglobin [Hb]) is present, and tissue iron storage is visible microscopically.
Platelet, neutrophil, and white blood cell counts fall.
Coagulation tests (bleeding time), reflecting decreased platelet count, are abnormal.
Bone marrow aspiration from several sites may yield a “dry tap,” and biopsy will show se verely hypocellular or aplastic marrow, with var ied amounts of fat, fibrous tissue, or gelatinous replacement; absence of tagged iron (because iron is deposited in the liver rather than bone marrow) and megakaryocytes (platelet precur sors); and depression of erythroid elements.
Differential diagnosis must rule out paroxysmal nocturnal hemoglobinuria and other diseases in which pancytopenia is common.
TREATMENT
Effective treatment must eliminate any identifiable cause and provide vigorous supportive measures, such as packed RBCs, platelets, and experimental histocompatibility locus antigen-matched leukocyte transfusions. Even after elimination of the cause, recovery can take months. Bone marrow or stem cell transplantation is the treatment of choice for anemia due to severe aplasia and for patients who need constant RBC transfusions. (See Bone marrow transplantation, page 466.)
Patients with low leukocyte counts need special measures to prevent infection. The infection itself may require specific antibiotics; however, these aren’t given prophylactically because they tend to encourage resistant strains of organisms. Patients with low Hb levels may need respiratory support with oxygen in addition to blood transfusions.
For older patients, or for those who don’t have a matched bone marrow donor, antithymocyte globulin (ATG) is an alternative treatment. ATG is a horse serum that contains antibodies against human T cells. It may be used in an attempt to suppress the body’s immune system, allowing the bone marrow to resume its blood cellgenerating function. Other immunosuppressant agents, such as cyclosporine, may also be used.
Other treatments may include corticosteroids to stimulate erythroid production, marrowstimulating agents such as androgens (which remain controversial), and colony stimulation factors to encourage growth of specific cellular components.
SPECIAL CONSIDERATIONS
If the platelet count is low (less than 20,000/µl), prevent bleeding by avoiding I.M. injections, suggesting the use of an electric razor and a soft toothbrush, humidifying oxygen to prevent drying of mucous membranes, avoiding enemas and rectal temperatures, and promoting regular bowel movements through the use of a stool softener and a proper diet to prevent constipa tion. Also, apply pressure to venipuncture sites
until bleeding stops. Detect bleeding early by checking for blood in urine and stool, and assessing skin for petechiae.
Take safety precautions to prevent falls that could lead to prolonged bleeding or hemorrhage.
Help prevent infection by washing your hands thoroughly before entering the patient’s room, by making sure he’s receiving a nutritious diet (high in vitamins and proteins) to improve his resistance, and by encouraging meticulous mouth and perianal care.
Watch for life-threatening hemorrhage, infection, adverse effects of drug therapy, or blood transfusion reaction. Make sure routine throat, urine, nose, rectal, and blood cultures are done regularly and correctly to check for infection. Teach the patient to recognize signs of infection, and tell him to report them immediately.
If the patient has a low Hb level, which causes fatigue, schedule frequent rest periods. Administer oxygen therapy as needed. If blood transfusions are necessary, assess for a transfusion reaction by checking the patient’s temperature and watching for the development of other signs and symptoms, such as rash, hives, itching, back pain, restlessness, and shaking chills.
Reassure and support the patient and his family by explaining the disease and its treatment, particularly if he has recurring acute episodes. Explain the purpose of all prescribed drugs and discuss possible adverse effects, including which ones he should report promptly. Encourage the patient who doesn’t require hospitalization to continue his normal lifestyle, with appropriate restrictions (such as regular rest periods), until remission occurs.
To prevent aplastic anemia, monitor blood studies carefully in the patient receiving anemia-inducing drugs.
Support efforts to educate the public about the hazards of toxic agents. Tell parents to keep toxic agents out of the reach of children. Encourage people who work with radiation to wear protective clothing and a radiationdetecting badge, and to observe plant safety precautions. Those who work with benzene (solvent) should know that 10 parts per million is the highest safe environmental level and that a delayed reaction to benzene may develop.
Bone marrow transplantation
In bone marrow transplantation, usually 500 to 700 ml of marrow is aspirated from the pelvic bones of a human leukocyte antigen (HLA)-compatible donor (allogeneic) or of the recipient himself during complete remission (autologous). The aspirated marrow is filtered and then infused into the recipient in an attempt to repopulate the patient’s marrow with normal cells. This procedure has effected long-term, healthy survival in about 50% of patients with severe aplastic anemia. Bone marrow transplantation may also be effective in patients with acute leukemia, certain immunodeficiency diseases, and solid tumor neoplasms.
Because bone marrow transplantation carries serious risks, it requires strict adherence to infection control and strict sterile techniques, and a primary nurse to provide consistent care and continuous monitoring of the patient’s status.
Before transplantation
Explain that the success rate depends on the stage of the disease and an HLA-identical match.
After bone marrow aspiration is completed under local anesthetic, apply pressure dressings to the donor’s aspiration sites. Observe the sites for bleeding. Relieve pain with analgesics and ice packs, as needed.
Assess the patient’s understanding of bone marrow transplantation. If necessary, correct any misconceptions about this procedure and provide additional information, as appropriate. Prepare the patient to expect an extended hospital stay. Explain that chemotherapy and possible radiation treatments are necessary to destroy cells that may cause the body to reject the transplant.
Various treatment protocols are used. For example, I.V. cyclophosphamide maybe used with additional chemotherapeutic agents or total body irradiation. This treatment requires aggressive hydration to prevent hemorrhagic cystitis. Control nausea and vomiting with an antiemetic, such as ondansetron or metoclopramide, as needed. Give allopurinol, as prescribed, to prevent hyperuricemia resulting from tumor breakdown products. Because alo pecia is a common adverse effect of high-dose cyclophosphamide therapy, encourage the patient to choose a wig or scarf before treatment begins.
Total body irradiation may follow chemotherapy, inducing total marrow aplasia. Warn the patient that cataracts, GI disturbances, and sterility are possible adverse effects.
Assess venous access. If necessary, the patient may have an indwelling central venous catheter inserted.
During marrow infusion
Monitor vital signs every 15 minutes.
Watch for complications of marrow infusion, such as pulmonary embolus, hypersensitivity reactions, and volume overload.
Reassure the patient throughout the procedure.
After the infusion
Continue to monitor the patient’s vital signs every 15 minutes for 2 hours after infusion, then every 4 hours. Watch for fever and chills, which may be the only signs of infection. Give prophylactic antibiotics as prescribed. To reduce the possibility of bleeding, don’t administer medications rectally or I.M.
Administer methotrexate or cyclosporine, as prescribed, to prevent graft-versus-host (GVH) reaction, a potentially fatal complication of allogeneic transplantation. Watch for signs or symptoms of GVH reaction, such as maculopapular rash, pancytopenia, jaundice, joint pain, and generalized edema.
Administer vitamins, steroids, and iron and folic acid supplements, as appropriate. Administration of blood products, such as platelets and packed red blood cells, may also be indicated, depending on the results of daily blood studies.
Provide good mouth care every 2 hours. Use hydrogen peroxide and nystatin mouthwash or oral fluconazole, for example, to prevent candidiasis and other mouth infections. Also provide meticulous skin care, paying special attention to pressure points and open sites, such as those from the marrow aspiration and I.V. insertion.
Sideroblastic anemias
Sideroblastic anemias are a group of heterogenous disorders with a common defect; they fail to use iron in hemoglobin (Hb) synthesis, despite the availability of adequate iron stores. These anemias may be hereditary or acquired; the acquired form, in turn, can be primary or secondary. Hereditary sideroblastic anemia may respond to treatment with pyridoxine. Correction of the secondary acquired form depends on the causative disorder; the primary acquired (idiopathic) form, however, resists treatment and usually proves fatal within 10 years after onset of complications or a concomitant disease.
CAUSES AND INCIDENCE
Hereditary sideroblastic anemia appears to be transmitted by X-linked inheritance, occurring mostly in young males; females are carriers and usually show no signs of this disorder.
The acquired form may be secondary to ingestion of or exposure to toxins, such as alcohol and lead, or to certain drugs, such as isoniazid used to treat tuberculosis. It can also occur as a complication of other diseases, such as rheumatoid arthritis, lupus erythematosus, multiple myeloma, tuberculosis, and severe infections.
The primary acquired form, known as refractory anemia with ringed sideroblasts, is most common in elderly people. It’s commonly associated with thrombocytopenia or leukopenia as part of a myelodysplastic syndrome.
In sideroblastic anemia, normoblasts fail to use iron to synthesize Hb. As a result, iron is deposited in the mitochondria of normoblasts, which are then termed ringed sideroblasts. (See Ringed sideroblast, page 468.)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


