Figure 12-1 Coagulation cascade. Solid arrows indicate activation and dashed lines indicate additional substrates activated by factor VIIa or thrombin. aPTT, activated partial thromboplastin time; HMWK, high molecular weight kininogen; PL phospholipid; PT, prothrombin time; TF, tissue factor.
DIAGNOSIS
Clinical Presentation
History
- A detailed history can assess bleeding severity, congenital or acquired status, and primary or secondary hemostatic defects.
- Prolonged bleeding after dental extractions, circumcision, menstruation, labor and delivery, trauma, or surgery may suggest an underlying bleeding disorder.
- Strong family history may suggest an inherited bleeding disorder.
Physical Examination
- Primary hemostasis defects are suggested by mucosal bleeding and excessive bruising.
- Petechiae: <2-mm foci of subcutaneous bleeding that do not blanch with pressure and typically present in areas subject to increased hydrostatic force: the lower legs and periorbital area (especially after coughing or vomiting)
- Ecchymoses: >3-mm black-and-blue (or violaceous) patches due to rupture of small vessels from trauma
- Petechiae: <2-mm foci of subcutaneous bleeding that do not blanch with pressure and typically present in areas subject to increased hydrostatic force: the lower legs and periorbital area (especially after coughing or vomiting)
- Secondary hemostasis defects can produce hematomas (localized masses of clotted/unclotted blood), hemarthroses, or delayed bleeding after trauma or surgery.
Diagnostic Testing
Laboratories
The history and physical exam guide test selection: Initial studies should include platelet count, prothrombin time (PT), activated partial thromboplastin time (aPTT), and peripheral blood smear.
Primary Hemostasis Testing
- A low platelet count requires a review of a blood smear to rule out platelet clumping artifact (often due to the EDTA additive), giant platelets, and misclassification of other cells as platelets.
- The bleeding time (BT) measures time until bleeding cessation from a standardized skin incision, but it does not quantify the perioperative risk of bleeding.2 While the BT is used to detect qualitative platelet defects, it is considered a poor test due to low accuracy and high interoperator variability. The platelet function analysis (PFA)-100 now replaces BT in most laboratories.
- The PFA-100 instrument assesses vWF-dependent platelet activation in flowing citrated whole blood. Most patients with von Willebrand disease (vWD) and qualitative platelet disorders have prolonged closure times. Anemia (hematocrit <30%) and thrombocytopenia (platelet count of <100 × 109/L) can cause artifactually prolonged closure times.
- In vitro platelet aggregation studies measure platelet secretion and aggregation in response to platelet agonists (e.g., adenosine diphosphate [ADP], collagen, arachidonic acid, and epinephrine), and they assist with the diagnosis of qualitative platelet disorders.
- Laboratory evaluation of vWD is discussed later in this chapter.
Secondary Hemostasis Testing
- Prothrombin time (PT): Measures time to form a fibrin clot after adding thromboplastin (tissue factor and phospholipid) and calcium to citrated plasma.
- Sensitive to deficiencies of the extrinsic pathway (factor VII), common pathway (factors X and V and prothrombin), and fibrinogen.
- Sensitive to anticoagulation therapy with warfarin and may also be prolonged with the use of direct thrombin inhibitors (DTIs) and factor Xa inhibitors.
- Reporting a PT ratio as an international normalized ratio (INR) reduces interlaboratory variation for monitoring warfarin therapy.3
- Sensitive to deficiencies of the extrinsic pathway (factor VII), common pathway (factors X and V and prothrombin), and fibrinogen.
- Activated partial thromboplastin time (aPTT): Measures the time to form a fibrin clot after activation of citrated plasma by calcium, phospholipid, and negatively charged particles.
- Sensitive to deficiencies of the intrinsic pathway (factors VIII, IX, and XI), common pathway, and fibrinogen and to clinically insignificant deficiencies of the contact activators (factor XII, prekallikrein, and high molecular weight kininogen).
- Sensitive to anticoagulation therapy with unfractionated heparin and may also be prolonged with use of direct thrombin inhibitors (DTIs) and factor Xa inhibitors.
- Although the aPTT may be prolonged with use of low molecular weight heparin (enoxaparin), monitoring of therapeutic anticoagulation should be performed with the anti–factor Xa inhibitor assay.
- Sensitive to deficiencies of the intrinsic pathway (factors VIII, IX, and XI), common pathway, and fibrinogen and to clinically insignificant deficiencies of the contact activators (factor XII, prekallikrein, and high molecular weight kininogen).
- Thrombin time: Measures time to form a fibrin clot after addition of thrombin to citrated plasma. Quantitative and qualitative deficiencies of fibrinogen, elevated fibrin degradation products, heparin, and DTIs may prolong the thrombin time.
- Fibrinogen: The addition of thrombin to dilute plasma and the measurement of a clotting time determine the effective functional concentration of fibrinogen. Conditions causing hypofibrinogenemia include decreased hepatic synthesis, massive hemorrhage, and disseminated intravascular coagulation (DIC).
- D-dimers result from plasmin digestion of fibrin. Elevated D-dimer concentrations occur in many disease states, including acute venous thromboembolism, DIC, trauma, and malignancy.
- General workup of unexpected prolonged PT or aPTT should include the following:
- Consideration of preanalytic variables such as incomplete filling of sample tubes, heparin contamination (screen with thrombin time), high hematocrit (>55%), hemolysis, and lipemia.
- A mixing study (performing PT or aPTT on the patient’s plasma mixed 1:1 with normal pooled plasma) will differentiate factor deficiencies from inhibitors:
- Correction of PT or aPTT to within reference range after mixing: simple deficiency of factor(s), depending on whether PT and/or aPTT is prolonged. Hemophilias, acquired deficiencies, and warfarin result in this pattern.
- No or minimal correction: factor inhibitor, depending on whether PT and/or aPTT are prolonged. Autoantibodies and alloantibodies to factors result in this pattern, as do heparins, DTIs, and lupus anticoagulants.
- Correction of PT or aPTT to within reference range after mixing: simple deficiency of factor(s), depending on whether PT and/or aPTT is prolonged. Hemophilias, acquired deficiencies, and warfarin result in this pattern.
- Common causes of PT and aPTT prolongation (Table 12-1):
- PT and aPTT prolonged: hypofibrinogenemia, common pathway factor (II, V, X) deficiencies, DIC, liver disease, dilutional coagulopathy, DTIs, factor Xa inhibitors, and superwarfarin (e.g., brodifacoum) toxicity
- PT only: warfarin, vitamin K deficiency, liver disease, factor VII deficiency
- aPTT only: Heparin, common hemophilias (factor VIII, IX, or XI deficiency)
- Most common inhibitors that prolong the aPTT are factor VIII inhibitors (if bleeding is present) and lupus anticoagulants (if there is no history of bleeding)
- PT and aPTT prolonged: hypofibrinogenemia, common pathway factor (II, V, X) deficiencies, DIC, liver disease, dilutional coagulopathy, DTIs, factor Xa inhibitors, and superwarfarin (e.g., brodifacoum) toxicity
- Consideration of preanalytic variables such as incomplete filling of sample tubes, heparin contamination (screen with thrombin time), high hematocrit (>55%), hemolysis, and lipemia.
TABLE 12-1 Factor Deficiencies Causing Prolonged Prothrombin Time (PT) and/or Activated Partial Thromboplastin Time (aPTT) That Correct with 50:50 Mix

PLATELET DISORDERS
Quantitative Platelet Disorders (Thrombocytopenia)
- Thrombocytopenia is defined as a platelet count of <140 to 150 × 109/L at most laboratories. In the absence of qualitative platelet defects or vascular damage, spontaneous bleeding often does not occur until the platelet count is <10 to 30 × 109/L.
- Thrombocytopenia occurs from decreased production, increased destruction, or sequestration of platelets (Table 12-2). Many infectious diseases are associated with thrombocytopenia through complex or poorly understood mechanisms.4
TABLE 12-2 Classification of Thrombocytopenia
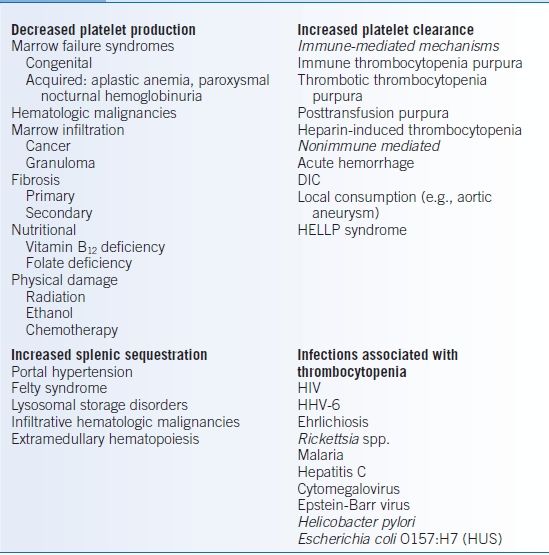
DIC, disseminated intravascular coagulation; HELLP, Hemolysis (with microangiopathy), Elevated Liver enzymes, and Low Platelet count; HUS, hemolytic uremic syndrome.
Immune Thrombocytopenic Purpura
GENERAL PRINCIPLES
Immune thrombocytopenic purpura (ITP) is an acquired autoimmune disorder in which antiplatelet antibodies cause shortened platelet survival and suppress megakaryopoiesis, leading to thrombocytopenia and increased bleeding risk. ITP can be classified into primary (idiopathic) and secondary (associated with coexisting conditions, including systemic lupus erythematosus, antiphospholipid antibody syndrome, HIV, hepatitis C, Helicobacter pylori, and lymphoproliferative disorders).4
DIAGNOSIS
Clinical Presentation
- ITP typically presents as mild mucocutaneous bleeding and petechiae or thrombocytopenia discovered incidentally.
- Primary ITP often has the scenario of isolated thrombocytopenia in the absence of a likely underlying causative disease or medication.
Diagnostic Testing
- Laboratory tests do not confirm the presence of primary ITP, though they help to exclude some secondary causes.
- Serologic tests for antiplatelet antibodies do not help diagnose ITP because of poor sensitivity and low negative predictive value and should not be used routinely.5
- Bone marrow biopsy and aspirate studies are not necessary in patients with typical presentations of ITP, irrespective of age. However, if patients have other abnormalities identified in history, physical examination, or peripheral blood count, bone marrow biopsy may be needed to exclude other causes such as malignancy, especially in older patients (age >60 years) and patients who do not respond to standard ITP treatments.6
- All newly diagnosed adult ITP patients should be tested for HIV and HCV.
TREATMENT
- The decision to treat primary ITP depends upon the severity of thrombocytopenia and symptoms of bleeding.
- Management of secondary ITP includes treatment of the underlying disease and standard primary ITP therapy.
- Initial therapy, when indicated, consists of glucocorticoids (typically prednisone 1 mg/kg/day for 2 to 3 weeks then taper depending on the patient’s responses and stability of platelet count).
- Steroid nonresponders or patients with active bleeding often also receive intravenous immunoglobulin (IVIG) (1 g/kg × 2 days) or anti–D immunoglobulin (WinRho) if Rh positive. Anti–D immunoglobulin is ineffective postsplenectomy.
- Approximately 80% of patients will respond to glucocorticoids initially with resolution of thrombocytopenia within 1 to 3 weeks.
- Steroid nonresponders and the 30% to 40% of patients who relapse during a steroid taper have chronic ITP. The therapeutic goals in refractory and relapsed ITP patients include a safe platelet count (>30 × 109/L) and minimization of treatment-related toxicities.
- Treatment options for refractory or relapsed ITP: The optimal sequence or choice of the following three treatment options has not been established and is usually determined by patient preference/conditions.
- Splenectomy: Two-thirds of patients with refractory ITP will obtain a durable complete response following splenectomy. Administer pneumococcal, meningococcal, and Haemophilus influenzae type B vaccines at least 2 weeks before splenectomy.
- Rituximab, an anti-CD20 monoclonal antibody, or much less commonly, other immunosuppressive agents such as cyclosporine, azathioprine, or androgen therapy with danazol.6,7
- Thrombopoietin (TPO) receptor agonists:
- Two small-molecule TPO receptor agonists are available for refractory ITP patients with increased bleeding risk. Romiplostim is given subcutaneously once weekly and eltrombopag orally once a day.
- Both produce durable platelet count improvements in 80% to 90% of refractory ITP patients after 5 to 7 days.
- Potential complications include transaminitis, thromboembolic events, and bone marrow fibrosis.8
- Two small-molecule TPO receptor agonists are available for refractory ITP patients with increased bleeding risk. Romiplostim is given subcutaneously once weekly and eltrombopag orally once a day.
- Splenectomy: Two-thirds of patients with refractory ITP will obtain a durable complete response following splenectomy. Administer pneumococcal, meningococcal, and Haemophilus influenzae type B vaccines at least 2 weeks before splenectomy.
Drug-Induced Thrombocytopenia
GENERAL PRINCIPLES
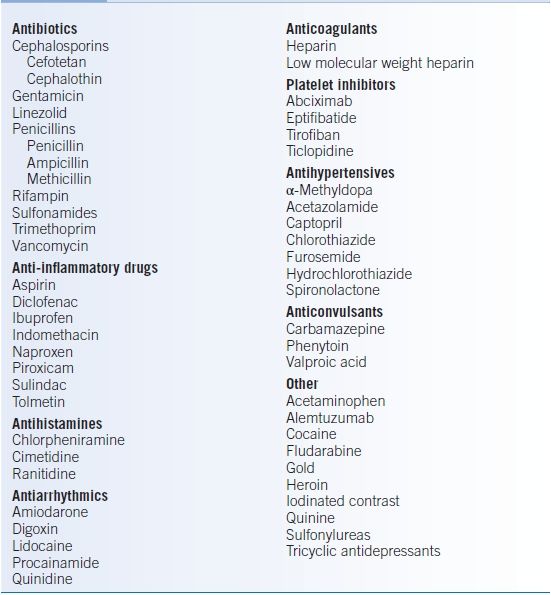
Drug-dependent immune thrombocytopenia results from drug-platelet interactions prompting antibody binding.9 Medications that are commonly associated with thrombocytopenia are listed in Table 12-3.10 An extensive list of medications with references can be found on the website of the University of Oklahoma Health Sciences (http://www.ouhsc.edu/platelets/DITP.html, last accessed January 2, 2015).
TABLE 12-3 Drugs Implicated in Immune Thrombocytopenia
DIAGNOSIS
Key factors to diagnose drug-induced thrombocytopenia:
- Exposure of suspected drug(s) precede(s) thrombocytopenia.
- Normalization of platelet counts with discontinuation of suspected drug(s).
- No other likely drugs present.
- No other more likely etiologies for thrombocytopenia.
- Thrombocytopenia recurs when patients are rechallenged with the same drug.
TREATMENT
- Discontinuation of the suspected offending agent(s) is the main therapy.
- Platelet transfusion for severe thrombocytopenia may decrease the risk of bleeding, but is discouraged for less severe thrombocytopenia without bleeding. IVIG, steroids, and plasmapheresis have uncertain benefits.
Thrombotic Thrombocytopenic Purpura
GENERAL PRINCIPLES
- The pathogenesis of thrombotic thrombocytopenic purpura (TTP) is the formation of disseminated platelet thrombi. Deficiency of the vWF-cleaving metalloprotease ADAMTS13 (most commonly autoantibody mediated) leads to elevated levels of abnormally large vWF multimers that spontaneously adhere to platelets and form occlusive vWF-platelet aggregates in the microcirculation and subsequent thrombotic microangiopathy (TMA).11 Thrombocytopenia due to platelet consumption, microangiopathic hemolytic anemia (MAHA) due to platelet microthrombi, and organ ischemia are characteristic features.
- Sporadic (primary) TTP has an incidence of approximately 11.3 cases per 106 people and occurs more frequently in women and African Americans.12
- Secondary TTP refers to TMA associated with DIC, HIV, malignant hypertension, vasculitis, organ and stem cell transplant–related toxicity, adverse drug reactions, and, during pregnancy, preeclampsia/eclampsia and HELLP (Hemolytic anemia, Elevated Liver enzymes, Low Platelet count) syndrome.
- TMA may also be associated with certain drugs, including quinine, ticlopidine, calcineurin inhibitors (cyclosporine, tacrolimus), sirolimus, and chemotherapy agents such as gemcitabine and mitomycin C.
DIAGNOSIS
Clinical Presentation
- The classic pentad of TTP includes consumptive thrombocytopenia, MAHA, fever, renal dysfunction, and fluctuating neurologic deficits, but is only fully present in <30% of cases. The findings of thrombocytopenia and MAHA alone should raise suspicion for TTP.
- Patients with autosomal recessive inherited ADAMTS13 deficiencies have recurrent TTP (Upshaw-Schulman syndrome).
Diagnostic Testing
- Schistocytes and thrombocytopenia on peripheral blood smears are classical findings.
- Hemolysis workup is often positive including anemia, elevated reticulocyte count, low haptoglobin, elevated lactate dehydrogenase, and elevated indirect bilirubin.
- TTP is often associated with very low or undetectable ADAMTS13 enzyme activity and an ADAMTS13 inhibitory antibody. However, TREATMENT should not be delayed for such testing, due to the urgency of the disease.
Treatment
- The mainstay of therapy is plasma exchange (PEX), requiring emergent inpatient admission Glucocorticoids (prednisone 1 mg/kg/day orally or methylprednisolone 1 g IV daily) are commonly initiated along with PEX. Treatment for refractory or relapsed TTP may include rituximab, cyclosporine, or vincristine.13–16
- Platelet transfusion in the absence of severe bleeding is relatively contraindicated because of the potential risk of additional microvascular occlusions.
- When TMA is suspected to be due to drugs, discontinuation of the offending agent is the preferred therapy. PEX is usually not effective.
Hemolytic-Uremic Syndrome
GENERAL PRINCIPLES
- The clinical pentad of TTP can also be found in hemolytic-uremic syndrome (HUS), but HUS is usually associated with higher incidence of renal dysfunction and lower incidence of neurologic manifestations.
- There are two types of HUS, typical and atypical.
- Typical HUS (diarrhea associated) often follows acute infection with Escherichia coli (especially serotype O157:H7) or Shigella dysenteriae that produce Shiga-like toxins.
- Atypical HUS (aHUS) has the clinical presentation of HUS without being preceded by the above-mentioned infections or diarrhea. Acquired and inherited defects in regulation of the alternative pathway of complement activation are frequently identified.17
- Typical HUS (diarrhea associated) often follows acute infection with Escherichia coli (especially serotype O157:H7) or Shigella dysenteriae that produce Shiga-like toxins.
DIAGNOSIS
Clinical Presentation
- Diarrhea (often bloody) and abdominal pain often precede typical HUS, and more pronounced renal dysfunction occurs.
- Familial atypical HUS often leads to chronic renal failure.
Diagnostic Testing
- Features of TMA and acute renal failure are usually present.
- In typical HUS, stool culture for E. coli O157 has a higher sensitivity than Shiga toxin assays.18
- ADAMTS13 activity is often normal or only mildly decreased (in contrast to TTP, which is associated with a very low ADAMTS13 activity).
- Workup for suspected aHUS should include molecular analysis of complement regulator factor H and I and MCP (membrane cofactor protein) genes as well as analysis for acquired inhibitors through reference laboratories.
TREATMENT
- Inpatient admission is recommended based on the severity of the presentation.
- Typical HUS:
- Treatment is supportive care (e.g., hydration).
- Antibiotics do not hasten recovery or minimize toxicity for HUS-associated infection.
- PEX is not recommended due to a demonstrated lack of benefit.
- Treatment is supportive care (e.g., hydration).
- Atypical HUS:
- PEX can provide some benefit, but is often less effective than for TTP.
- Eculizumab, a monoclonal antibody against the complement protein C5, inhibits the complement-mediated renal injury.
- PEX can provide some benefit, but is often less effective than for TTP.
Heparin-Induced Thrombocytopenia
GENERAL PRINCIPLES
- Heparin-induced thrombocytopenia (HIT) is an acquired hypercoagulable disorder caused by antibodies targeting heparin and platelet factor 4 (PF4) complexes, which can activate platelets, cause thrombocytopenia, and increase thrombin generation.19
- HIT typically presents with a decreased platelet count by at least 50% after exposure to unfractionated heparin (UFH) or (less likely) low molecular weight heparin (LMWH).
- The incidence of HIT varies with clinical setting, anticoagulant formulation, dose, duration of exposure, and previous exposure, ranging from 0.1% to 1% in medical and obstetric patients receiving prophylactic and therapeutic UFH to 1% to 5% in patients receiving prophylactic UFH after total hip or knee replacements or cardiothoracic surgery.20
DIAGNOSIS
Clinical Presentation
- Suspect HIT when thrombocytopenia occurs during heparin exposure by any route in the absence of other causes of thrombocytopenia and when platelet counts recover after cessation of heparin.
- HIT usually develops between 5 and 14 days of heparin exposure (typical-onset HIT). Exceptions include delayed-onset HIT, which occurs after stopping heparin, and early-onset HIT, which starts within the first 24 hours of heparin administration in patients with recent exposure to heparin.20
- Both venous and arterial thromboembolic complications occur due to activation of platelets. Thrombosis can precede, be concurrent with, or follow recognition of thrombocytopenia. Thrombi may occur at heparin injection sites as full-thickness skin infarctions, sometimes in the absence of thrombocytopenia. HIT rarely causes severe thrombocytopenia (<20 × 109/L) and bleeding.
Diagnostic Testing
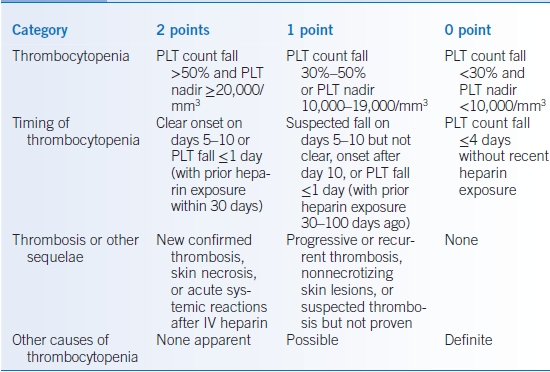
- A scoring system based on the “4Ts” improves diagnostic accuracy: thrombocytopenia, timing, thrombosis, and other explanations for thrombocytopenia (Table 12-4).21 A 4 T score of 3 or below correlates with a low probability of HIT and high negative predictive value (0.998).22 A HIT assay should NOT be ordered due to the high false-positive rate in this population.
- There are two main types of HIT assays:
- Serologic enzyme immunoassays (PF4 EIA) to detect heparin-PF4 antibodies
- Functional assays: platelet aggregometry or serotonin release assay to detect activation of control platelets in the presence of patient serum and heparin
- Serologic enzyme immunoassays (PF4 EIA) to detect heparin-PF4 antibodies
- In comparison, serologic tests have higher sensitivity and serve as screening tests, whereas functional assays have higher specificity and may serve as confirmatory tests when the diagnosis is unclear.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


