- All cells in a multicellular organism must coordinate their behavior and subordinate their individual requirements for the overall good of the organism. This is only possible because cells have a variety of ways to communicate with one another and an elaborate machinery that allows them to interpret and integrate often diverse messages into responses (e.g. grow–don’t grow; live–die).
- The size of an organism and constituent tissues is to a large extent dictated by cell number and is subject to a variety of often draconian measures, which ensure tissue homeostasis by counterbalancing cell division and survival with the opposing processes of cell death and senescence.
- The size of the adult organism is largely genetically determined, but for many organs, size is far from immutable. In fact, numerous adult tissues exhibit a remarkable degree of plasticity and can adapt to changes in the environment, such as nutrient availability, or changing demands of the organism.
- The actual means by which “appropriate organ size” is monitored by the organism and, if deemed necessary, cell number and size adjusted are poorly understood, but require the coordination not only of cellular growth and survival but often also cell adhesion, pattern formation, and polarity. The whole can usefully be thought of as an “organ-size checkpoint,” analogous to the cell-cycle checkpoints discussed in Chapter 4. The interestingly named “Hippo pathway” is emerging as a strong contendor for the role of organ size controller.
- Secreted factors that promote or inhibit growth allow adaptive changes in tissue mass and can originate from local cells (paracrine) or more distant ones that release hormones into the circulation (endocrine).
- Growth factors influence cellular replication by binding to specific receptors on, or occasionally within, the cell to form active complexes that in turn unleash a cascade of downstream signaling events. In general, sequential activation of kinase enzymes by addition of phosphate groups eventually carries the growth message to the nucleus, where it triggers expression of genes needed for cell cycle. Growth factors are critically important in coercing cells to leave G1 and enter S phase of the cell cycle – the so-called G1/S transition.
- As the signal passes onwards, the previous participants are inactivated by phosphatases that remove the added phosphate groups, readying the system for the next stimulus. Signaling cascades are rarely if ever linear, despite the often misleading appearance given by diagrams in textbooks (including this one), and fall subject to a bewildering array of inhibitory inputs and convergence points with other pathways as well as negative feedback and positive feedforward loops.
- Not surprisingly, such convoluted pathways offer numerous opportunities for epimutations to aberrantly stimulate growth or remove normal growth-inhibiting restraints. Fortunately, however, we have in-built protective mechanisms that will prevent unalloyed growth resulting from any single mutation; the quotidian apoptosis and senescence will effectively cancel out the aberrant growth-promoting effect – a molecular embodiment of Newton’s third law.
- By implication, normal cell replication requires that mitogenic signals be accompanied by those that prevent apoptosis or senescence, even if these derive from the same growth factor.
- Tumors are highly complex structures concocted of often multiple distinct subpopulations of cancer cells, endothelial cells, and stroma and imbued with the paraphernalia of chronic inflammation.
- It is often said that cancers are forged in the fire of chronic inflammation, steeped in growth-promoting factors and cytokines, but thereafter are burnished by mutations that generate growth signals from within. Mutations can activate growth factor signaling at various different levels including:
- enhanced production of growth factors, either directly by the cancer cells themselves or indirectly by stimulating their production by other cells in the microenvironment,
- increased expression of growth factor receptors (particularly the receptor tyrosine kinases),
- expression of constitutively active receptors that trigger downstream signaling even in the absence of a ligand,
- activation of downstream signaling pathways directly or indirectly by loss of a normal negative feedback loop or inhibitory protein,
- deregulated expression or activity of transcription factors that promote cell cycle.
- enhanced production of growth factors, either directly by the cancer cells themselves or indirectly by stimulating their production by other cells in the microenvironment,
- Of the growth-promoting signaling pathways, the overlapping RAS–RAF–ERK and PI3K–AKT pathways are among the most pivotal as they transduce mitogenic and growth or survival signals, respectively. They also illustrate the schemata described above. Thus, most human cancers have activating mutations at one or more of the following levels:
- growth factor receptor tyrosine kinases (RTK), such as HER2 and epidermal growth factor receptor (EGFR), are constitutively activated or upregulated,
- RAS may become activated by loss of inherent or facilitated inactivating GTPase activity,
- BRAF mutations can activate signaling downstream of RAS,
- loss of tumor suppressors, such as NF-1 and PTEN, can contribute to aberrant activation of the pathway,
- transcription factors, such as Myc, may be deregulated.
- growth factor receptor tyrosine kinases (RTK), such as HER2 and epidermal growth factor receptor (EGFR), are constitutively activated or upregulated,
- It is rare for redundant mutations to coexist, so in general either RAS or RAF are mutated not both. Moreover, mutations in either will remove the requirement for EGFR activation and render such cancer cells unresponsive to EGFR inhibitors.
- Growth factors are not usually oncogenes or produced in excess as a direct result of mutations, although some tumor cells produce and secrete growth factors. In many cases growth factors are synthesized by nurturing stromal cells or as part of inflammatory processes that often underlie the development of a carcinoma.
- In order to activate signaling pathways, growth factors usually require integrin-mediated cell adhesion. This ensures activation only occurs when cells are in their correct environment.
- Two highly cancer-relevant processes, epithelial–mesenchymal transition (EMT) and formation of cancer stem cells, as well as the signaling pathways that regulate them, are being explored as new drug targets.
Introduction
So divinely is the world organized that every one of us, in our place and time, is in balance with everything else.
Johann Wolfgang von Goethe
The size of an organism or organ is determined by its total cell mass – in other words, the total number of cells and their size. Four overlapping processes regulate cell mass and the control points in these processes are the preeminent targets for cancer-causing mutations:
- cell growth,
- cell division,
- cell differentiation, and
- cell death.
These processes usually combine together in a well-orchestrated and exquisitely balanced series of performances throughout ontogeny and adult life, ensuring that everything is in the right place at the right time and of the appropriate size. The responsibility for interpreting and conducting the repertoire rests with the specific and shared regulatory factors and signaling pathways that will be discussed in this chapter. The duration and timing of the performances is of central importance – an overenthusiastic conductor leading an unending series of encores will result in overgrowth and cancer.
The signals regulating cell size and cell number, originating from distant or neighboring cells, activate appropriate responses by “docking” with specific cell surface receptors or enter the cell to bind proteins inside the cell. Not all such extracellular signals are soluble secreted proteins but also include proteins bound either to the surface of other cells or to components of the extracellular matrix (ECM). Moreover, these can operate in various different ways and combinations to regulate cell division and number as shown in Table 5.1.
Table 5.1 Some regulatory processes affecting cell proliferation
| Growth factor dependence | Cell proliferation requires tissue-type specific mitogenic and survival factors, the absence of which may also trigger apoptosis. Cells are also exposed to a variety of growth-inhibitory factors such as TGF-β |
| Anchorage dependence | Cell proliferation requires transmembrane proteins called integrins to interact with components of the extracellular matrix (ECM) – again such contacts may be required to prevent apoptosis |
| Contact inhibition | Contact with like cell types inhibits cell movement and proliferation |
| Senescence | Observations of vertebrate somatic cells in culture have identified a limitation on the number of times a cell can divide (around 50–70 divisions for human cells) before the cells cease further division – the Hayflick limit. This is difficult to examine in the intact organism, although loss of telomeres, which is a major factor in determining the Hayflick limit, is also clearly a mediator of replicative arrest in vivo. What is clear is that senescence can be coupled to signals that regulate replication and, together with apoptosis (cell death), may act as a counterbalance, particularly to inappropriate or excessive activation of mitogenic signals within the cell |
| Apoptosis | Many mitogenic signals intracellularly are also coupled to pathways, which can induce apoptosis |
| Nutrient and oxygen supply | Expansion of cell numbers may result in a tissue outgrowing its blood supply and could result in deprivation of oxygen (hypoxia) or nutrients or accumulation of damaging waste products, all of which might in turn limit further tissue growth. Moreover, the endothelial cells lining the inside of the vessels may secrete survival factors also contributing to regulation of tissue growth. Thus, cells may undergo apoptosis or necrosis unless tissue growth is accompanied by new vessel development or expansion (angiogenesis) |
All of these processes are also inherent barriers to neoplasia and must be overcome in order for tumors to form and progress.
The factors regulating cell mass are conveniently divided into three classes based on their predominant cellular action, although individual factors may regulate more than one process.
- mitogens promote cell division largely by allowing G1/S transition in the cell cycle;
- growth factors promote an increase in cell mass (cell growth) by enhancing protein synthesis;
- survival factors promote cell survival by suppressing apoptosis.
Students should be aware that the term “growth factor” (GF) is frequently employed to describe a protein that has any of these properties – as evidenced by the names by which almost all of them are known.
Platelet-derived growth factor (PDGF) was among the first proteins found to have a mitogenic action. Originally described as a serum mitogenic factor, PDGF was subsequently identified as the product of platelets released during blood clotting. Now more than 50 different protein mitogens have been identified with different degrees of selectivity for certain cell types. In addition, there are factors, exemplified by members of the transforming growth factor-β (TGF-β) family, which also inhibit cell replication (cytostatic). Many factors have properties additional to those on cell division and can promote cell growth, survival, or motility. Table 5.2 gives a description of some key growth factors.
Table 5.2 A short list of some known growth factors
Once a given signaling process has been triggered, the message is propagated and disseminated within the cell in order to ultimately elicit some kind of response, which in the context of cancer might be to divide or become motile. In most cases, proteins involved in signaling cascades activate their targets transiently by joining together and then apart. Interactions require collisions and often result in altered phosphorylation, with the whole facilitated by having the interacting participants arranged in close proximity within the cell – a kind of molecular Morris dance.
In this chapter we will commence by identifying the links between growth factors and the cell cycle before discussing general concepts of growth and differentiation. We will then outline the normal regulation of cell proliferation by growth factors, the receptors through which they act, the downstream signaling pathways activated by these receptors, and finally the activation of nuclear transcription factors, which regulate genes involved in cell cycling (see Fig. 5.13 for a simple schematic of growth factor signaling). As all of these steps involve proteins whose deregulated activity has been shown to contribute to cancer (in particular the oncogenes), we will proceed to discuss how such processes become derailed during cancer formation.
Over the last few years, there has been an explosive increase in knowledge of the key roles played by microRNAs (miRNAs) in posttranscriptional control of gene expression (see Chapters 7 and 11), which is impacting essentially all areas of cell biology. Signaling pathways, which are often exquisitely sensitive to dose effects, appear to be particularly sensitive to miRNA-mediated regulation and this has been recently shown for epidermal growth factor (EGF), TGF-β, and WNT. Finally we will conclude by discussing how knowledge of growth factor signaling has been exploited to develop a new generation of targeted anticancer agents, including the tyrosine kinase inhibitors (TKIs). Signaling pathways feature in every chapter, but to avoid duplication a detailed description of a few key proteins and their upstream and downstream partners feature in other chapters. Thus, RAS, MYC, and SRC are described in Chapter 6 (oncogenes), survival pathways in Chapter 8 (cell death), tumor suppressors and DNA damage in Chapters 7 and 10, and cell adhesion and motility in Chapter 12. Drug treatments based on growth signaling are covered in Chapter 16.
It is worth noting here that signaling pathways are often described and illustrated as linear chains or isolated cascades, with activation of an upstream protein triggering activation of one or more proteins down the chain and so on. This is a simplification to aid understanding. Most steps have critical regulatory inputs, including not just negative and positive feedback loops but also extensive crosstalk between pathways. After all, it is highly improbable that cells will be exposed to growth factors individually. It is much more likley that cells are continually having to simultaneously integrate any number of different extracellular stimuli. In fact, crosstalk and integration is a way by which complexity of responses can result through the presence of key nodes, as exemplified by the G1/S transition, where the cell must reach a “decision” based on integrating all the information being delivered to it. As will be seen, the result of crosstalk can be to aggregate a number of weak, below-threshold signals into a response or for one mutually antagonistic signal to shut off another if sufficiently strong. Moreover, by focusing on concentration and amplitude of a signal, the importance of dynamics, such as rates of change, oscillations, and frequency, to signaling may be lost and foster the often erroneous impression that cell fate is solely determined by proteins fixed in an active or inactive conformation.
So we should appreciate that dynamics of oscillations, as opposed to simply amplitude (as has been demonstrated for the NF-κB pathway) may generate different cell responses, even if for now we file away that knowledge for future reference. After all, with an estimated 150 000 bona fide binary protein interactions alone in the human protein universe we have our work cut-out just to determine the members and sequence of entire functional protein networks.
Another issue often clouded in diagrams of signaling pathways relates to how proteins that occupy adjacent positions in a signaling cascade ever manage to get together within the comparatively vast and fluid volume of the cell. It is little use if an activated protein never manages to bump into a target protein in order to propagate the signal. Imagine throwing grains of rice around inside a gothic cathedral. How often will they collide with one another? Clearly, steps must be taken to maximize the chances of such interactions. Well, one way would be to flick the grains along the floor or upon a single tile space, thus increasing the probabilities of a “collision” by constraining molecules to an effectively two-dimensional playing field. An alternative would be to line them along the pillars from floor to ceiling, which could also ensure that the direction of travel is anatomically apt. The cellular counterparts of this would be localizing prenylated proteins to the inner aspect of the cell membrane and the lining up of signaling molecules along cellular scaffolds, such as cytoskeletal elements from surface to nucleus.
These issues notwithstanding, for most purposes the simplified model of signaling is sufficient, as it allows us to understand the context of cancer-causing mutations within functional categories and also to unravel and predict how new and existing drugs alter cell behavior. In the future we may also learn how to target cancer signaling by interrupting localization (the rationale behind farnesyl transferase inhibitors to impede RAS signaling) and even to alter dynamics.
Growth Factor Regulation of the Cell Cycle
People are like earth. They can either nourish you and help you grow as a person or they can stunt your growth and make you wilt and die.
Plato
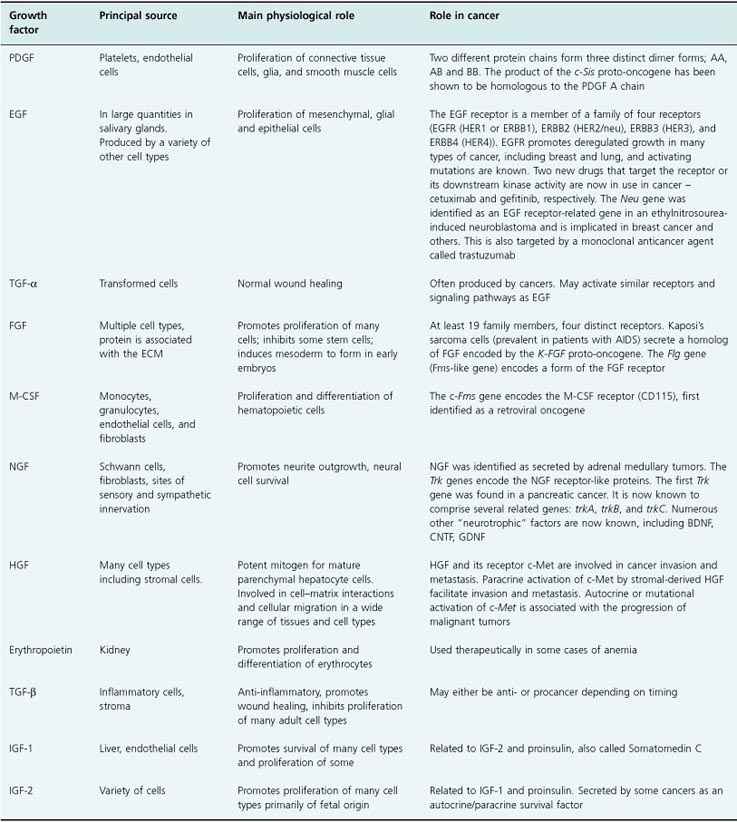
Cells in early embryos replicate very rapidly (a mere half hour) and can start DNA replication as soon as mitosis is completed. This is due to the absence of gap phases G1 and G2 (normally present in cycling cells of the adult; see Chapter 4), and likely serves the needs for rapid expansion of cell numbers following fertilization. However, later in development and particularly in the adult, this replication potential is restrained and subject to tighter controls. As discussed in the previous chapter, the adult cell cycle contains two gap periods: G1 phase between M (nuclear division) and S (DNA synthesis) and a G2 phase between S and M. Not only do these gaps allow for a period of monitoring and correction of any DNA damage or errors that may have arisen during DNA replication, but critically G1 also provides the opportunity for many diverse signals to be integrated into a decision as to whether the cell will actually replicate, pause, or indeed remain alive. To a degree, a similar scenario applies to terminally differentiated cells in G0, seeking to re-enter the cell cycle (Fig. 5.1). Importantly, it is increasingly apparent that any given cell is probably exposed simultaneously to a variety of stimulatory and inhibitory signals, arising locally and via the circulation (Fig. 5.2), all of which have to be integrated by that cell before it decides whether to replicate. This integration is performed by complex interacting networks of intracellular signals generated in response to these extrinsic regulating factors. It is the net balance of all these responses that determines whether a cell enters the cell cycle (Fig. 5.3).
Figure 5.1 Growth factors act particularly at the G1 phase of the cell cycle. (a) The point at which cells commit to replicate their DNA and enter the cell division cycle is controlled by the RB protein, the product of the tumor suppressor gene RB. RB acts as an “off switch” for entry into S phase by binding the transcription factor E2F required for expression of S-phase genes. After appropriate growth factor stimulation, the inhibitory effects of RB are removed by phosphorylation, which allows E2F to activate S-phase genes during early G1 required for cell cycle progression. (b) A special case for differentiated cells re-entering cell cycle. Blocks to replication in “terminally differentiated” quiescent cells are somewhat different to those generally described for the regulation of cell cycle in other cells, probably because less is known, but clearly show some overlap. Three blocks to cell cycle progression in “quiescent cells” are shown in thick black lines, which can be overcome by different mitogenic stimuli. The mid-G1 block is overcome by cyclin D–CDK4 activity induced by growth factor-regulated genes such as c-MYC. Viral proteins such as E1A and T antigen are not susceptible to these blocks and are capable of pushing previously quiescent cells through one or more cell cycles, but then result in apoptosis, which restrains oncogenic potential. E2F or cyclin E–CDK2 expression alone cannot overcome the late G1 block, while cyclin D1 and CDK4 alone cannot take the cycle beyond G2.
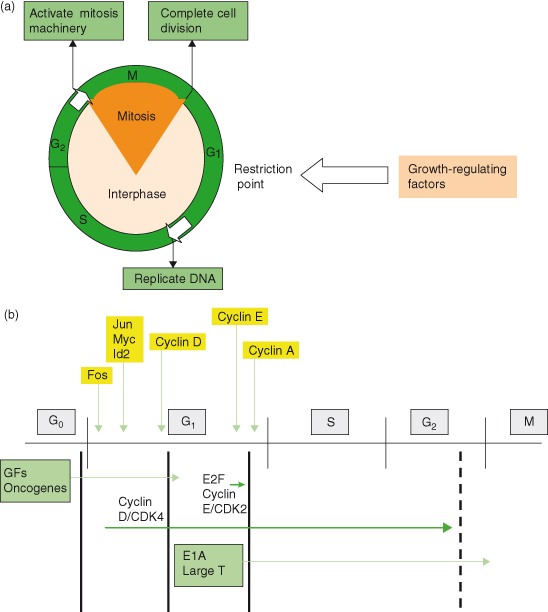
Figure 5.2 Growth-regulatory factors derive from local and circulating sources. Factors regulating cellular replication and survival include secreted growth factors as well as cell–cell and cell–matrix interactions. Growth factors may be produced locally by the cancer cell or cells of a different type and act by an autocrine (on the same cell) or paracrine (on a neighboring cell) route. Growth factors can be delivered via the circulation (endocrine) or be secreted from the vascular endothelial cells or by stromal cells. Stromal cells can also produce MMPs that can act to liberate growth factors from matrix or can directly affect production of ROS and thus DNA damage in the cancer cell. Recruited immune cells, including activated T lymphocytes and macrophages may also produce growth-regulating cytokines.
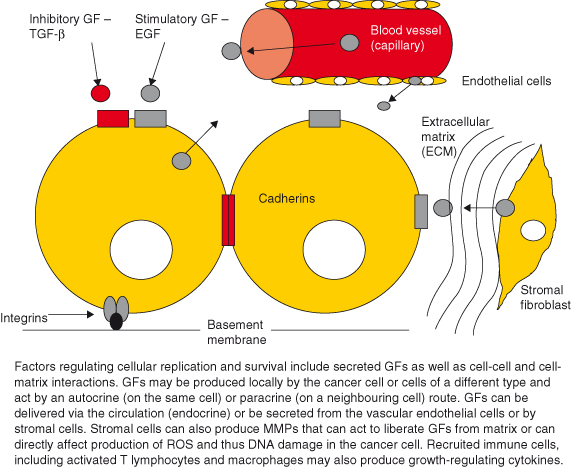
Figure 5.3 Cell division depends on the balance of different growth-regulating signals. Ultimately, the decision whether a cell lives or dies, replicates, or does not is dependent on the balance of positive and negative signals received.
Growth factor signaling systems are now believed to operate as complex networks rather than the traditional view of signaling as linear pathways. Complex signaling networks enable integration of multiple inhibitory and stimulatory inputs into binary choices by the cell, including: replicate or arrest; survive or die. Moreover, there are numerous sites of overlap between signals regulating progression through the cell cycle and those that can permanently prevent a cell from replicating by promoting either senescence or even cell death.
Although there is considerable overlap and crosstalk, broadly two of the most important signaling pathways activated by various growth factors are:
- the mitogenic RAS–RAF–ERK (MAPK) cascade and a key transcription factor, c-MYC (Fig. 5.4) and
- the PI3K–AKT survival pathway.
Figure 5.4 Main signaling pathways for mitogens. Mitogenic factors largely, but not exclusively, act via RTK to activate in particular the MAPK/ERK cascade.
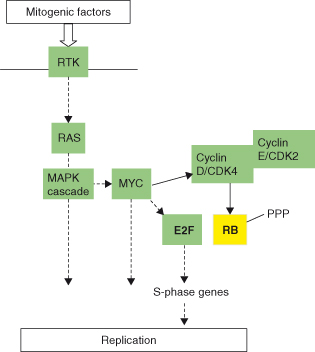
Together, these pathways may act synergistically to allow cell-cycle entry and progression while also avoiding various failsafe mechanisms seeking to trigger cell death and growth arrest. Not surprisingly, RAS, c-MYC (see Chapter 6) and various activators of the PI3K–AKT survival pathway are frequently involved in cancer and will often feature in subsequent chapters.
Mammalian cells must pass through a restriction point, or “R” point (equivalent of the start point in yeast cells), before committing themselves to entering the S phase and duplicating their entire DNA. Importantly, once the cell cycle has progressed beyond this point (G1/S transition) it will proceed even if the mitogenic stimuli are removed. The net result of many positive and negative growth-regulating signals normally determines whether a cell makes the G1/S transition and proceeds to DNA replication, and, unsurprisingly, such controls are usually lost in cancer cells. Diverse growth-regulating factors operate largely at the level of activation of cyclin-dependent kinases (CDKs), which drive the “cell cycle engine.” Prominent among these regulatory factors are the polypeptide growth factors, which either promote or inhibit the G1/S transition (Fig. 5.5).
Figure 5.5 Positive and negative growth regulators determine G1/S transition and cell cycling. SMAD and FOXO together mediate inhibitory action of TGF-β on cell replication by activating CDK inhibitors (CKI). In contrast, growth factors and c-MYC act to promote cell replication by upregulating cyclin D and inhibiting activation of CKIs such as p15INK4b and p27KIP1.
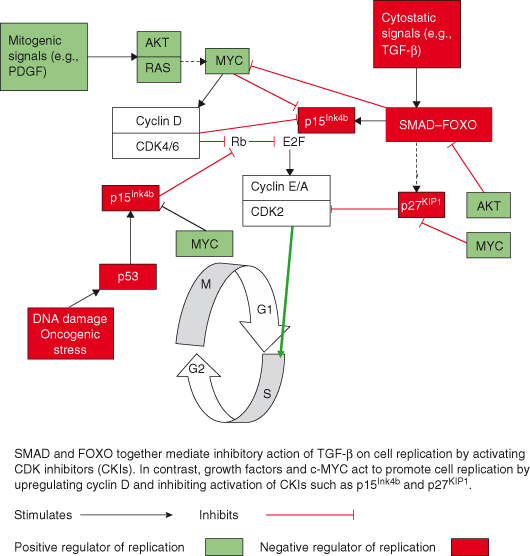
Factors that promote the cell cycle (mitogens) generate signals resulting in increased activity of cyclin D–CDK holenzyme complexes during the G1 phase, which, as described in Chapter 4 (Fig. 4.2), promote phosphorylation and inactivation of the tumor suppressor retinoblastoma (RB) protein a prerequisite for releasing the brakes on the cell cycle and allowing expression of S-phase genes. Conversely, inhibitory “cytostatic” signals, in particular the transforming growth factor-β (TGF-β), among other actions, activate inhibitors of CDK4, such as p15INK4b, thus preventing RB phosphorylation. Cytostatic signals may also activate two other CDK inhibitors (CKIs) (see Chapter 4), p21Cip1 and p57Kip2.
It is also important to reiterate here that apart from controlling cell growth and cell cycle, some growth factors also activate signaling pathways that affect other cellular processes including cell survival, migration, and differentiation – cellular processes that if disrupted inappropriately are important in the behavior of the cancer cell (see Table 5.1).
Growth Homeostasis and Tissue Repair and Regeneration
In every animal which has not passed the limit of its development, a more frequent and continuous use of any organ gradually strengthens, develops and enlarges that organ, and gives it a power proportional to the length of time it has been so used; while the permanent disuse of any organ imperceptibly weakens and deteriorates it, and progressively diminishes its functional capacity, until it finally disappears.
Jean-Baptiste Lamarck
Multicellular organisms, like human societies, rely on the cooperative behavior of their constituents. This is particularly important with respect to ensuring that cell numbers, functions, and localization are appropriately maintained. In general, such control over cell behaviors becomes increasingly stringent as the organism completes its development – the majority of cells sacrificing a large part of their potential to replicate and becoming more specialized in their functions. Thus, cells stop cycling and differentiate to give rise to populations of cells that exhibit specialized functions, such as skin, nervous system, etc. Importantly, within the adult organism, cells repress their innate potential to grow and divide beyond their normal boundaries and contrary to patterns dictated by the overall developmental plan of the organism. This can be a particularly tricky business given that the rules normally restraining these behaviors can be relaxed when required for wound healing, regeneration of damaged tissue, or to allow adaptive growth of a particular tissue or cell population to changing demands (e.g. growth and modulation of breast tissue during lactation; growth of uterine tissue during the menstrual cycle) (Fig. 5.6).
Figure 5.6 Restraints on cell replication and motility may be temporarily released. During injury to tissues cells may be allowed to replicate and migrate, but this is transient.
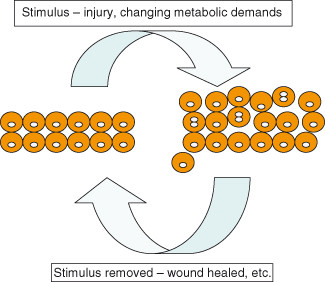
Regenerative and adaptive capacity differs greatly across organs and organisms. Under such circumstances, many cells may be given a degree of freedom to expand numbers and to migrate into what is for them an unusual location, where they would not normally be allowed. The major challenge, given the obvious similarities in behavior to those of cancer cells, is to ensure that enhanced cell replication and motility are temporary. Therefore, when the tissue in question has returned to its normal state (e.g. the wound has healed, or lactation has ended), the normally strict controls are reapplied successfully. A simple, clear example of such a case is given in Chapter 12 (see Box 12.4).
Regenerative processes that involve unleashing unrestrained cellular migration and replication carry with them an inherent risk of cancer that is counterbalanced by several restraining processes. Important among these are limiting the extent and duration of regenerative capacity (note the cancer risk of chronic inflammation) by initiating and terminating signals, and tight control over stem cells, dedifferentiation, transdifferentiation, and proliferation and patterning.
Thus, in multicellular organisms, cellular cooperation is normally ensured by numerous extrinsic and intrinsic factors, which together regulate cell growth, proliferation, motility, and survival in order to best serve the requirements of the whole organism (see Fig. 5.2). It is not surprising then that disruption of the tight controls normally imposed on these processes in the adult can have drastic effects on cells and tissues of the body, leading to diseases such as cancer. In fact, one of the common defining features of cancer is that of uncontrolled and unscheduled cellular proliferation; cancer cells divide relentlessly and are able to resist the usual mechanisms regulating replication in normal cells. Normal cells require external growth factors (mitogens) to divide – at least in order to make the G1/S transition. When synthesis of these growth factors is inhibited or access restricted by some other means, then cells stop dividing. Normal cells also respond to growth-inhibiting signals, such as TGF-β.
One of the “hallmark” features of cancer is autonomous cell division, such that cancer cells are no longer dependent on such positive or negative growth factors. Cell behavior is not just regulated by secreted factors but also by molecules on the surface of neighboring cells, such as integrins and adhesion molecules (see Chapter 12). Normal cells show contact inhibition in vitro; that is, they respond to contact with other cells by ceasing cell division. This characteristic is lost in cancer cells, as demonstrated in cell culture where cancer cells continue to divide and “pile up” to form transformed colonies in vitro (see Chapter 6 and Fig. 6.3).
Normal cells also age and die, and are then replaced in a controlled and orderly manner by new cells when possible. It is widely accepted that there are limits on the number of times that normal cells can divide (at most 50 times in cell culture) before they exit permanently from cell division either by becoming senescent or by dying. This intrinsic cell division “counter” is operated at least in part by the progressive shortening of structures at the ends of chromosomes called telomeres; a process referred to as “telomere attrition.” In cancer cells, the enzyme involved in maintaining telomeres, telomerase, is reactivated and this contributes to continued cell divisions. Cellular senescence may also be promoted by mechanisms involving tumor suppressors such as RB (see Chapters 7 and 9). Lastly, normal cells will cease to divide and undergo apoptosis when there is DNA damage too extensive to be repaired (see Chapters 8 and 10), or when cell division is abnormal. In contrast, cancer cells continue to divide under these conditions (e.g. in cells that have lost p53), enabling the propagation of cells with abnormal DNA. The signaling pathways activated by these various factors show considerable overlap (Fig. 5.7).
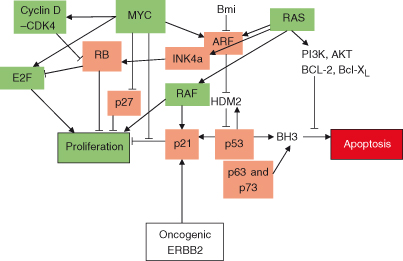
Figure 5.7 Cooperation between oncogenic signaling pathways that regulate cell replication, and apoptosis. Activation of RAS and MYC results in potential engagement of both replication and growth but also of apoptosis and possibly growth arrest. MYC triggers G1/S transition and cell cycle entry by regulating expression of cyclin D and CDK4 (upregulated) and p21CIP1 and p27KIP1 (downregulated). Together with cyclin E–CDK complexes (not shown), the net effect is hyperphosphorylation of RB and release of E2F for transcription of S-phase genes. If either MYC or RAS levels are excessive (as might occur during oncogenesis) or other proapoptotic signals are received, then the balance may be tipped away from replication. RAS can promote senescence through either p16INK4a or ARF, which activate RB or p53 pathways respectively, while MYC can inhibit growth arrest/senescence by inhibiting p21CIP1 and inducing TERT (telomerse reverse transcriptase). RAS may also activate p21CIP1 via RAF activation. Although MYC may activate the apoptotic pathway (e.g. via ARF), RAS is able to suppress apoptosis by activating the PI3K pathway and, subsequently, AKT. It can readily be appreciated how oncogenic MYC and RAS may conspire in oncogenesis. The combination of RAS and MYC acting together provides a potential means of avoiding apoptotic and growth arrest mechanisms, activated by either acting individually. Moreover, it can also be appreciated how inactivating mutations in RB or p53 may collaborate in tumorigenesis by enabling the cancer cell to avoid either growth arrest or apoptosis or both.
An Organ-Size Checkpoint
One of the great challenges for modern biology was to unravel exactly how an organism determines that a given tissue is of the correct size (or for that matter how an individual cell determines how big it is). A checkpoint, analogous to those described in Chapter 4, has been proposed; this “organ-size checkpoint” in some way determines the size of an organ and, if required, triggers corrective processes that will adjust cell size and number. Broadly, organism or tissue size is regulated through balancing of positive and negative growth regulatory signals within cells.
Much remains to be learned about this fundamental process but studies in fruit-flies and nematode worms have revealed some basic principles. First, nutrient signals are important players, and, possibly via hormones such as insulin and insulin-like growth factors (discussed later), these activate intracellular signaling pathways that regulate both cell size and replication/survival. One protein activated by such signaling, mTOR (mammalian target of rapamycin), is of particular interest and is discussed later. Very important regulatory processes also involve cell death control. The two most important in this context are (1) activity of proteins such as c-MYC which can activate both cell replication/growth on the one hand and also apoptosis on the other and (2) a particular form of apoptosis, termed anoikis, which is triggered in cells that have inadvertently left their usual tissue location, have breached a tissue boundary, and have wandered into an inappropriate location (see Chapter 8). Finally, recent studies suggest that the organization of a tissue plays an important role in controlling cell growth. Thus, cellular polarity (many cells, particularly in epithelia, usually face in a particular direction – “up or down”) and interactions between neighboring cells, basement membranes, and extracellular matrix all influence cellular behavior, including differentiation, replication, and survival (see Chapter 12).
Specific signaling pathways other than those traditionally linked to replication and survival have been identified as important in determining size. These include the aforementioned mTOR, but also the regulators of stem cells and “stemness” in cancer cells (Wnt, Notch, and Hedgehog pathways) and the conserved Hippo tumor suppressor pathway (all discussed in more depth later in the chapter).
Regulated and Deregulated Growth
There are some trees, Watson, which grow to a certain height and then suddenly develop some unsightly eccentricity. You will see it often in humans. I have a theory that the individual represents in his development the whole procession of his ancestors, and that such a sudden turn to good or evil stands for some strong influence which came into the line of his pedigree. The person becomes, as it were, the epitome of the history of his own family.
Sir Arthur Conan Doyle, The Adventure of the Empty House
In the living intact organism changes in cell number reflect the balance of cell proliferation and cell death, so that an increase in tissue mass can result from either suppression of cell death (apoptosis) or increase in proliferation. However, processes regulating cell volume or size are increasingly appreciated as independent contributors to cell or tissue mass (see Box 5.1).
Self-evidently, in order for oncogenic (cancer-causing) mutations (see Chapter 6) to give rise to cancer, the mutated cells must divide or else the mutation will not be propagated. It is widely accepted that cancers are most likely to originate from the most replicatively active cells, such as stem cells or progenitor cells (see Box 5.2). In part this reflects the increased probability of acquiring mutations when DNA is being copied during the cell cycle and also the fact that this will then be propagated. However, many other properties of stem cells resemble those found in cancer cells, such as longevity and self-renewal.
In general, the turnover of cells in most adult organs is slow, with small numbers of cells lost being replaced by replication of surviving cells of the same type or by neogenesis from stem cell precursors (Fig. 5.8) or even from cells of another type (transdifferentiation). Despite rapid progress in this area, it may be surprising to learn that there is still considerable debate in many situations as to the relative contributions of these various processes of cell renewal to normal tissue homeostasis and, moreover, to situations where more substantive cell renewal is required, such as after injury or during regeneration. During development, in general cell replication exceeds cell death and the organism grows, whereas in adult life the processes are balanced and tissue size is maintained. When mutations occur in nondividing cells, such as neurons, they rarely induce cancer, which accounts for the rarity of such tumors in adults. However, some tissues with usually low rates of turnover exhibit considerable plasticity during adult life; for example, many endocrine glands can undergo adaptive increases in mass in response to increased demands for their hormone output.
Figure 5.8 Tissue mass homeostasis. Tissue mass in the adult is maintained by balancing cell losses with cell renewal. Cells lost through apoptosis (necrosis or other cell death) or by shedding are normally replaced by replication of existing differentiated cells of the same, or occasionally even of another, cell type or by replication and differentiation of adult stem cells. Self-evidently if cell losses in any tissue exceed the ability for renewal then the tissue will shrink (good examples being loss of insulin-producing beta cells in human type 1 and type 2 diabetes), conversely if cell gains exceed losses then the tissue will expand (as exemplified in the initial response of beta cells to conditions demanding more insulin production). Many tissues are capable of switching between these conditions, thus enabling adaptation to demands – tissue plasticity. Some tissues are regarded as static, with effectively no turnover in the adult. Any losses in such tissues will result in progressive involution or atrophy of that tissue with time.
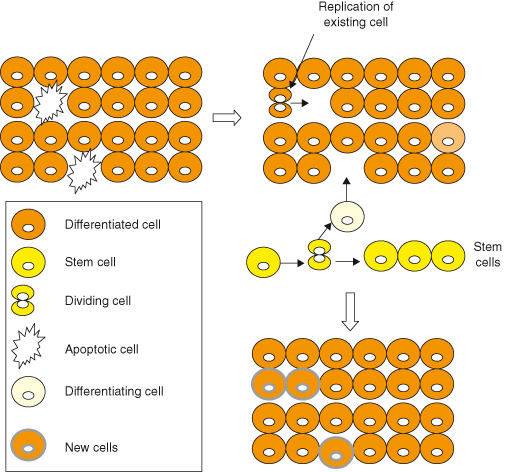
Importantly, such adaptive growth is not inevitably a response to a pathological process, but is a normal part of physiology, for example during pregnancy, where demands for various hormones such as insulin increase. Particularly in this latter scenario, adaptive growth of insulin-secreting pancreatic β cells is usually followed by β-cell apoptosis and involution of the previously expanded islet tissue post partum, when the demand for insulin is reduced. Intriguingly, recent studies employing a variety of tools for in vivo lineage-tracking suggest that both replication of pre-existing β cells and differentiation from progenitor cells or cells of another lineage contribute to β-cell mass regulation with preference determined by the nature of the damaging insult and relative availability of suitable precursors. Adaptive growth may also occur in response to nutrient deprivation or disorders in metabolism. Thus, the parathyroid glands may undergo hyperplasia (increase beyond normal cell mass) in response to reducing serum calcium (secondary hyperparathyroidism).
Importantly, under some circumstances, such as that following a prolonged or frequently repeated period of adaptive growth, hyperplasia of a tissue may become autonomous and continue even after the stimulus has been removed. A well-known example of this occurs in the parathyroid glands – a condition known as tertiary hyperparathyroidism. In other circumstances, hyperplasia may indeed progress to formation of a benign (adenoma) or malignant (adenocarcinoma) neoplasia. Presumably in these cases, the growth-promoting environment may also provide a fertile one for the accumulation of oncogenic mutations and for expansion of mutated cells. In this scenario, such oncogenic mutations (e.g. activated c-Myc or Ras; loss of p53 or Rb) would drive excessive cell replication independently of the normal regulatory signals (see Chapters 6 and 7).
Cancers occur in many tissues populated predominantly by nondividing differentiated cells. Stem cells are the major or only usual source of new cells in such tissues, which include epidermis and gut epithelium where cell losses are extensive (frequent mechanical damage, normal regular sloughing of cells from the surface), or when the normally minimal cell losses in a given tissue are increased after injury or by inflammation. It is generally believed that stem cells may be the major origin of cancers in these cases, though many recent studies have shown that terminally differentiated cells can readily become less differentiated and re-enter the cell cycle under certain circumstances and may even be induced to acquire stem cell characteristics (stemness) by undergoing EMT. Together this supports the notion that not all cancers necessarily arise in the stem cell compartment (discussed in more detail under cancer stem cells). In general, differentiation of a cell greatly reduces its replication potential. Under some circumstances, such as during inflammatory conditions, even differentiated epidermal keratinocytes may replicate, though this is associated with some loss of or delay in acquiring of the normal fully differentiated phenotype. The association between differentiation and replication is clearly seen in human tumors, with the most poorly differentiated cells (for example in the metastases) exhibiting the shortest cell division times.
Cellular Differentiation
The elementary parts of all tissues are formed of cells in an analogous, though very diversified manner, so that it may be asserted, that there is one universal principle of development for the elementary parts of organisms, however different, and that this principle is the formation of cells.
Theodore Schwann
The cells comprising the various tissues of a multicellular organism often exhibit dramatic differences in both structure and function. In fact, if you consider the differences between a mammalian nerve cell and a lymphocyte it becomes difficult to imagine that the two cells have identical genomes. In fact, for many years differentiation was held as evidence against the central dogma of genetics and indirectly contributed to the beginnings of what we know as epigenetics (see Chapter 11) – namely, information that can be passed on to the progeny during cell division not encoded in the DNA. Originally, it had been suggested that differentiation might actually involve the irreversible loss of some genes (DNA). This is now known not to be the case, rather differentiation involves the progressive alteration of gene expression. Various factors may produce the unique patterns of gene expression characteristic of any given cell type, including silencing of some genes by methylation and chromatin modification (epigenetics) and selective activation of others.
Importantly, it is increasingly apparent that many cell types do not irreversibly alter gene expression during differentiation, rather they may actually retain the ability to adopt a more primitive developmental state if required and stimulated by the appropriate signals or potentially under the influence of cancer-causing mutations or epigenetic alterations. In fact, it may be a normal feature of cells that retain the potential to replicate in the adult (and this does not just apply to stem cells), to “de-differentiate” somewhat in order to be able to replicate. This is well illustrated in cancer cells, which are highly replicative and almost invariably show some loss of the normal differentiated features of the cell of origin. Thus, in several tumor models activation of c-Myc may cause loss of differentiation alongside cell-cycle entry.
It has long been appreciated, and is being increasingly explained at a molecular level, that embryonic development, tissue injury and repair, chronic inflammation, and cancer all have many common features that distinguish them from normal tissues. It is not intended to give an exhaustive list here but rather to indicate some general principles. Common features include a greater replicative and motility potential of cells, increased presence of cytokines and growth factors, more cells undergoing EMT, and showing stemness and reactivation of many signaling pathways usually confined to ontogeny or adult stem cells. Two examples that show reactivation of signaling otherwise confined to development are described here:
- Re-expression of IGF-2 due to somatic epimutations has often been shown in rodent and human cancers. It is functionally important in some cancers and in extreme cases can be sufficiently raised to increase IGF-2 protein in the circulation and mimic the effects of insulin causing low blood glucose levels. Whether re-expression of IGF-2 might actually contribute to initiation of cancer has been addressed in some studies. It has been shown that loss of imprinting (LOI) of Igf2 (see also Chapter 11) in a mouse model doubles the risk of developing intestinal tumors and this is associated with a less differentiated phenotype of cells, even in the normal colonic mucosa. Similar observations have also been made in humans, with LOI of IGF2, suggesting that altered maturation of non-neoplastic tissue may be one mechanism by which epigenetic changes affect future cancer risk.
- The Hedgehog protein (Hh), which controls cell proliferation and differentiation in normal limb formation and bone differentiation, is also implicated in the self-renewal of stem cells in adult tissues. Persistent Hh pathway activation has been shown to contribute to various cancers, including some forms of skin cancer.
Differentiation Therapy
The rationale behind differentiation therapy is the assumption that cancer cells have been fixed in an immature or less differentiated state and that this contributes to accelerated replication and avoidance of apoptosis. Differentiation therapy aims to drive cancer cells back into a more differentiated state and resume the process of maturation. It is therefore not in itself cytotoxic but instead restrains growth and may sensitize the cells to potential apoptotic triggers or chemotherapy. The first successful differentiation agent was all-trans-retinoic acid (ATRA) used to treat acute promyelocytic leukemia (APL). APL is the result of a translocation (an exchange of chromosome material) between chromosomes 15 and 17, which results in formation of a chimeric protein comprising the promyelocytic leukemia protein (PML) and the retinoic acid receptor alpha (RARa), which regulates myeloid differentiation. PML–RAR causes an arrest of maturation in myeloid cells at the promyelocytic stage, contributing to expansion of promyelocytes. Treatment with ATRA causes the promyeloctes to differentiate and may result in remission rates of up to 70%.
The Concept of Terminal Differentiation
Traditionally, cells that have irreversibly lost the ability to proliferate during the process of developing specialized functions (differentiation) have been described as terminally differentiated. However, this term can be misleading because it implies that a given adult cell type cannot proliferate. In fact, many cell types previously believed terminally differentiated have subsequently been shown to be capable of proliferation; in the previous chapter the concept was introduced that some cells may remain quiescent (in G0) for extremely long periods of time waiting for an appropriate stimulus to re-enter the cell cycle. So in general it may be preferable to think about differentiated cells that are not replicating as quiescent unless one is absolutely certain. In some cases, of which skin keratinocytes are a notable example, cells lose their nuclei during terminal differentiation and are as a consequence unambiguously irreversibly growth arrested. However, such an obvious example aside, one must be cautious in adopting the definition of terminal differentiation, as has been done in the past, to all those cell types that are perceived to be unable to proliferate to any useful extent and therefore would not usually contribute to tissue growth, maintenance, or repair.
One illuminating example would be that of the insulin-secreting β cells of the pancreatic islets. In this case it was long accepted that the β-cell mass was maintained and underwent adaptive increases in growth through the replication of a progenitor cell or cells, but, recent studies using “lineage tracking” have clearly shown that β cells can themselves re-enter the cell cycle and are, surprisingly, the predominant usual source of new β cells in the adult. Crucial to resolving the seeming paradox, namely, that differentiation and replication are generally incompatible, has been the repeated demonstration that differentiated cells can shake off some of their specialized functions (and change gene and protein expression to more closely resemble that of an earlier developmental stage) – termed cell “plasticity” – in order to replicate. Figure 5.1 shows a schematic representation of how cell-cycle entry and progression may be regulated in a well-studied, usually quiescent cell type, the myotube.
In fact, in most cancer cells, at least some of the “markers” of the presumed mature cell type from which the cancer cell may have originated are absent or reduced (compared to the normal cell counterpart). This also reflects directly on an important question in cancer biology – what is the cell of origin for various cancers? This is addressed in other chapters, but is worth noting here. Because cancer cells are in general less differentiated, does this imply that they have originated in stem cells that may have “differentiated” a bit, or conversely in mature cells that may have “de-differentiated” a bit?
These intriguing questions aside, what is clear is that increasing or restoring differentiation (specialized behaviors) in cancer cells may be one means of reducing or even removing their replicative or invasive potential. Thus, understanding the mechanisms that cause permanent loss of proliferative capacity or at least prolonged quiescence in differentiated cells is of major interest and may lead to the design of new therapeutic strategies aimed at targeting differentiation of cancer cells. Differentiation is not the only means by which cells may lose their ability to proliferate, however, and replicative senescence, which may be induced by activation of tumor suppressor pathways is discussed in more detail in Chapter 9.
Tissue Growth and the “Angiogenic Switch”
During development and under some circumstances in the adult (e.g. wounding), tissue adaptation and regeneration are accompanied by the growth of new blood vessels, a process known as angiogenesis (see Chapter 14). The resultant supply of blood to the growing tissue – aside from delivering nutrients and oxygen and removing waste products and carbon dioxide – establishes an access route for circulating regulatory factors (endocrine route – Fig. 5.2) and immune cells (see Chapter 13). Importantly, it now seems increasingly likely that the vascular endothelial cells also provide growth factor signals that have a direct local influence on cell survival and growth of the tissue.
The notion that tissue expansion per se may be associated with the concurrent provision of an appropriate vasculature is not a novel one. For angiogenesis to take place, an appropriate increase in angiogenesis-promoting factors and or a decrease in angiogenesis-suppressing factors must occur. However, for many years based on landmark studies by Judah Folkman, Doug Hannahan, and others the prevailing view has been that during tissue expansion in cancer an “angiogenic switch” was required and probably conferred by a specific and distinct mutagenic event that enabled a would-be cancer to become angiogenic and thereby for cancer to progress. By implication, such a switch would activate angiogenesis-promoting factors or deactivate suppressors, thus enabling a tumor to become angiogenic and expand. However, although this may be the case in some cancers, recent results challenge the idea that this is a general requirement. Thus, tissue growth under the influence of some oncogenes, notably c-Myc and Ras, may automatically be associated with an angiogenic response without a further “oncogene driven switch.” In other words, the activity of these proteins includes activation of angiogenesis. In some cases this may relate to the development of a deficiency in oxygen supply (hypoxia) as a growing tissue begins to outstrip its existing blood supply or the direct secretion of angiogenic factors such as VEGF. A more detailed discussion of angiogenesis is given in Chapter 14.
Cancers and Nutrients
The cause of nutrition and growth resides not in the organism as a whole but in the separate elementary parts – the cells.
Theodore Schwann
As discussed above, cancer-causing epimutations ultimately enable cells to overcome environmental signals that normally regulate their behavior and to replicate and grow uncontrollably even to the detriment of the organism. Many cancer-causing epimutations involve deregulation of ancient regulatory pathways that are present in primitive organisms such as yeast, where they may largely respond to changes in environmental nutrient availability and coordinate decisions about growth and reproduction accordingly. In general, abundant nutrients activate mTOR, whereas nutrient-poor conditions favor activation of inhibitors of mTOR, such as the tumor suppressors tuberous sclerosis complex 1 (TSC1), TSC2, and liver kinase B1 (LKB1). Interestingly, inherited loss of any of these or of upstream inactivators of the RAS–RAF–ERK (MAPK) or PI3K–AKT pathways that otherwise activate mTOR, such as NF-1 or PTEN, produce clinical cancer syndromes with similar features (see Chapter 3).
Tumor Cell Metabolism – the “Metabolic Switch”
Tumor cells require a fuel supply that is able to support the increased replication and growth characteristic of a progressing cancer. Broadly, there are two main ways by which cells produce energy (as adenosine triphosphate – ATP): oxidative phosphorylation in the mitochondria and glycolysis in the cytoplasm. Although both processes are present in essentially all cells, oxidative phosphorylation predominates, with glycolysis increasing at times of oxygen deprivation, such as in exercising muscles.
In 1930, the German biochemist and Nobel Prizewinner Otto Warburg suggested that cancer might be caused by deranged energy processing in the cell. This ran counter to then (and largely still now) prevailing views that such changes are rather symptomatic of cancers rather than causes.
Warburg first described the reliance of tumors on glucose metabolism for energy nearly a century ago. In fact, the ability of tumors to switch from an aerobic metabolism of glucose within the tricarboxylic acid cycle to an anaerobic metabolism of glucose is essential for the progress and maintenance of many cancers, and Warburg showed that such cancers relied on glycolysis even in the presence of oxygen – the Warburg effect. Thus cancer cells use an altered metabolic program to that employed by normal differentiated adult cells; they take up more glucose and process it largely by aerobic glycolysis, producing large quantities of secreted lactate, ATP, water, and carbon dioxide. The Warburg effect thus provids substrates and energy for tumor growth because cancer cells take up increased amounts of glucose and channel it largely through aerobic glycolysis rather than oxidative phosphorylation. The Warburg effect is triggered by various oncogenes, such as c-Myc and can be arrested by tumor suppressors.
This metabolic switch is regulated by multiple signaling pathways (Fig. 5.9). Thus, tumor cells frequently upregulate proteins involved in glucose uptake, such as the glucose transporter GLUT-1 and various enzymes involved in anaerobic glucose metabolism, including hexokinase, required for initial phosphorylation of glucose to glucose-6-phosphate (G6P). G6P can then be further catabolized to produce energy or can be stored as glycogen. Tumor cells also upregulate enzymes involved in glycolysis. The M2 isoform of pyruvate kinase (PKM2) promotes the metabolism of glucose by aerobic glycolysis and contributes to anabolic metabolism. Phosphoenolpyruvate (PEP) is the usual substrate for pyruvate kinase and can act as a phosphate donor by phosphorylating phosphoglycerate mutase (PGAM1), a key glycolytic enzyme. Not surprisingly, then phosphorylated PGAM1 correlates with PKM2 expression in tumors. Interestingly, however, this alternative glycolytic pathway may actually be driven by reduced pyruvate kinase enzyme activity in PKM2-expressing tumor cells.
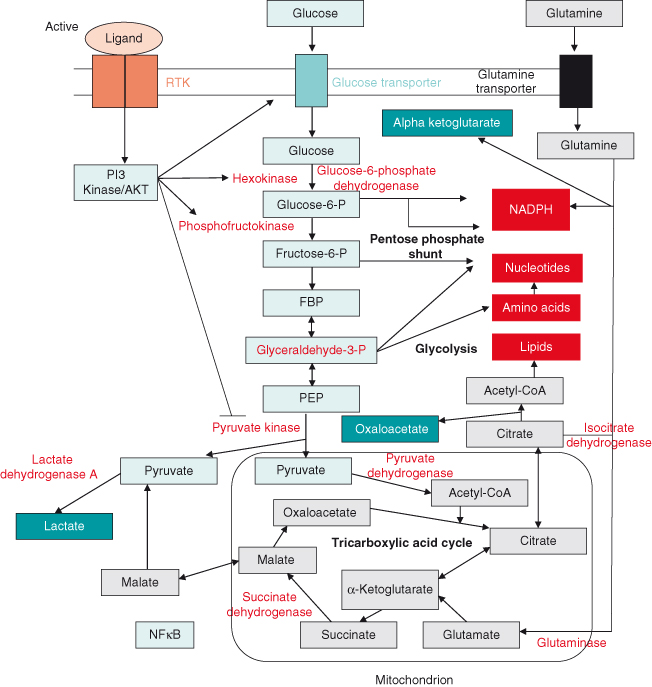
As intimated earlier, as tumors expand they often outstrip their oxygen supply (vascular supply) and become hypoxic. As a result, proteins such as HIF-1 are activated and may induce gene expression changes, including those regulating glycolysis. It remains to be shown whether glycolysis happens completely independent of hypoxia in cancer cells.
Warburg’s hypothesis is currently experiencing a renaissance and recent studies are suggesting that a shift in energy production from oxidative phosphorylation to glycolysis may be the seventh hallmark feature of cancer. By implication, if the Warburg effect is essential to cancer cells then novel targeted therapies might be designed to exploit this. Although Warburg attributed this process to defective mitochondrial function, it now seems more likely that instead it is a product of mutations in various signaling pathways, such as AKT and MYC, which can in various ways direct increased glucose uptake, which together might render the cancer cell independent of regulatory signals, such as insulin. Glycolysis is less efficient (produces two molecules of ATP for every glucose molecule, whereas complete oxidation produces 38), but increasing the rate of glycolysis on top of oxidative phosphorylation may easily provide extra energy supplies needed to drive proliferation and growth of cancer cells. Moreover, what may seem a rather poor and inefficient way to generate ATP (energy), is actually a potential route by which the cancer cell can accumulate key intermediates, such as nucleotides, amino acids, and lipids, needed to support growth and cell division. Enhanced glycolysis is also exploited in a variety of diagnostic procedures in clinical practice and underlies the ability of imaging techniques such as positron emission tomography (PET) to identify cancers, because these more avidly take up the radiolabeled glucose analog (see Chapter 2).
Several novel targeted treatments are under investigation based on the perceived importance of the Warburg effect for cancers – irrespective of whether this is secondary to hypoxia, and include glucose analogs, such as 2-deoxy-D-glucose, which can downregulate glycolysis, AKT inhibitors such as the asymmetric bisimidazoacridones, inhibitors of the proton pump v-ATPase, which acts to deacidify the cytosol in cancer cells, other metabolic targeting agents such as glufosfamide, and those which can block key glycolytic enzymes.
Growth Factor Signaling Pathways
Ships that pass in the night, and speak each other in passing, only a signal shown, and a distant voice in the darkness; So on the ocean of life, we pass and speak one another, only a look and a voice, then darkness again and a silence.
Henry Wadsworth Longfellow
Arguably, this is the single most important area in modern cancer biology and is the main focus of most initiatives aiming to develop new drugs, improved diagnostics, and deliver the holy grail of personalized medicine.
Growth factor signaling pathways affect essentially all biological processes and are particularly important in multicellular organisms, although many are highly conserved. Growth factor signaling is invariably disturbed in cancers, either by epimutations in key receptors or downstream signaling molecules which confer aberrant activation and sometimes deactivation of the pathways, thus unleashing some aspect of cancer behavior. The central role of signaling in cancer is emphasized by the fact that this and the following two chapters are largely concerned with nothing else.
The balance between cell death and survival or replication in the organism is determined in large part by growth factors that dictate whether a given cell will live or die. Epidermal growth factor (EGF) is a prototypic extracellular growth factor that regulates cell replication and survival through binding to cell surface receptor tyrosine kinases and activation of a network of signal transduction pathways, including the PI3K–AKT, RAS–MAPK–ERK, and JAK–STAT pathways. These pathways in turn activate or inhibit various transcription factors that regulate expression of genes/proteins involved in growth regulation. In cancer, such signaling pathways are among the most frequently deregulated, and in the case of EGF and family members, they are one of the most important targets for treatments.
The transmission and transduction of extracellular growth signals into the expression of genes involved in cell replication is fundamental to our understanding of cancer biology. Not surprisingly, most of the major cancer-causing mutations (but by no-means all) relate in some way to these signaling pathways, with mutations that either activate signaling molecules which promote growth (oncogenes – see Chapter 6) or disable molecules required for arresting cell replication or inducing apoptosis (tumor suppressors – see Chapter 7). Moreover, there is no shortage of signaling molecules to be deregulated – 539 kinases alone are encoded in the human genome.
Broadly speaking, proteins encoded by cancer-relevant genes can be ordered or classified according to the level they occupy within a growth factor signaling pathway and also by the nature of the key functional domains of the oncoprotein. One such scheme is outlined in Table 5.3 and will be followed in Chapter 6, where oncogenes are discussed in more detail.
Table 5.3 Classification of cancer-relevant genes
| Growth factors | Including platelet-derived growth factor (PDGF), epidermal growth factor (EGF), and vascular endothelial growth factor (VEGF) |
| Receptors | Can be either extra- or intracellular and are activated once bound by a suitable ligand usually originating from another source outside the cell |
| Receptor tyrosine kinases | The cell membrane receptors for various growth factors, such as PDGF and EGF, and oncogenes such as HER2/NEU are receptor tyrosine kinases (RTKs). In general, ligand binding leads to dimerization of receptors and activation on intrinsic tyrosine kinase activity that regulates downstream signaling. These tyrosine kinases have proved to be spectacularly amenable to therapeutic inhibition and are the target of a vast number of new anticancer agents – the tyrosine kinase inhibitors (TKIs) |
| G-protein coupled receptors | Including the angiotensin receptor |
| Nuclear receptors | Not all growth factors/extracellular ligands act via receptors on the cell surface, but some can enter the cell and bind to nuclear receptors. These are ligand-dependent transcription factors that regulate cell growth and differentiation in many target tissues and include receptors for steroid hormones, such as the estrogen receptor, and other ligands, such as the peroxisome proliferator-activated receptor (PPAR) family and farnesoid X receptor (FXR) |
| Signaling transducers and pathways | Primarily involving enzyme cascades that transduce, amplify, and allow crosstalk and integration of inputs from diverse sources. Most such enzymes activate or deactivate their target proteins, which may themselves be enzymes, by either adding or removing phosphate groups |
| Membrane-associated G-proteins |



