Chapter 9 Granulomatous, necrobiotic and perforating dermatoses
Sarcoidosis
Clinical features
Sarcoidosis (Gr. sarkos, flesh; eidos, form), so-named because its histological features were originally thought to resemble a sarcoma (Boeck), is a common systemic disease of unknown etiology. It is characterized and defined by the presence of noncaseating granulomata, usually (but not invariably) affecting multiple organ systems.1–10 Manifestations are variable. Patients may present with:
Sarcoidosis is more commonly encountered in industrialized countries and shows particularly high incidences in northern Europe (including the UK), the USA, and New Zealand, where as many as 20/100 000 of the population may be affected. It presents particularly in people in their third and fourth decades and shows a female predominance.11 In the USA, sarcoidosis is common among blacks and there is a similar tendency in the UK (Figs 9.1, 9.2). An epidemiological study of sarcoidosis in the Detroit, Michigan, area found that African-Americans living there had a 3.8 times greater risk of developing the disease compared with Caucasians.12 The disease is rare in Asians.13 First- and second-degree relatives of patients with sarcoidosis seem to have a significant risk of developing the disease compared to the normal population.14 The disease is rare in children, presents mainly in teenagers and although the manifestations are usually similar to those seen in adults, infants may present with symptoms simulating juvenile rheumatoid arthritis (Fig. 9.3).15–17 Two forms of sarcoidosis have been identified in children. A variant affecting older children and occurring mainly during teenage years presents with a multisystemic disease similar to that seen in adults. Younger children under the age of 4 present with a cutaneous rash, arthritis, and uveitis.18 Infantile sarcoidosis should not be confused with Blau’s syndrome. This disease is inherited in an autosomal dominant fashion and is characterized by sarcoidal granulomata in the skin, uveal tract and joints but with no pulmonary involvement.19,20 Despite the similarities between both diseases, no genetic linkage has been identified.
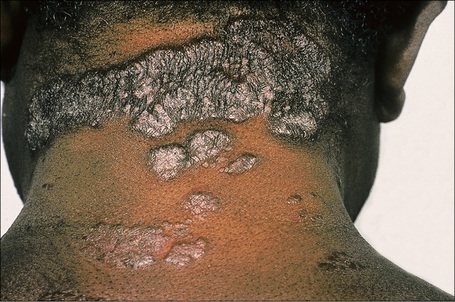
Fig. 9.1 Sarcoidosis: this patient presented with multiple plaques with raised margins on the neck.
From the collection of the late N.P. Smith, MD, the Institute of Dermatology, London, UK.
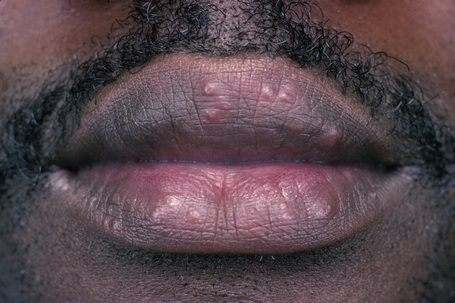
Fig. 9.2 Sarcoidosis: papules are present on both upper and lower lips.
From the collection of the late N.P. Smith, MD, the Institute of Dermatology, London, UK.
Rarely, sarcoidosis presents in monozygotic twins.21 Coexistence with common variable immune deficiency is also a rare occurrence.22
Cutaneous lesions occur in 20–35% of patients with systemic sarcoidosis and may be classified into non-specific (erythema nodosum) and specific (granulomatous) subtypes.10 Cutaneous sarcoidal granulomata appear to be associated with a poorer prognosis and an increased incidence of pulmonary fibrosis and uveitis. Chronic facial lesions have been shown to be more commonly associated with involvement of the lungs, sinuses, and eyes.23 Erythema nodosum occurs quite commonly in sarcoidosis, reported incidences varying from 11% to 31%.24 There is a significant female predominance (3:1). Interestingly, erythema nodosum appears to be relatively uncommon in both African-Americans and Caucasians in the USA. It presents as erythematous, tender, subcutaneous nodules, usually on the anterior tibial regions. Erythema nodosum may be associated with pyrexia, polyarthralgia (wrists, knees, and ankles), a very high erythrocyte sedimentation rate (ESR) and bilateral hilar lymphadenopathy (Lofgren’s syndrome). This acute form of sarcoidosis is associated with a good prognosis, with most patients experiencing resolution within 6 months of onset of symptoms.25–27 In one study, however, 16% of patients who presented with erythema nodosum developed chronic disease.26
A not uncommon mode of presentation is the development of a widespread, usually asymptomatic, maculopapular eruption. Individual lesions are erythematous or violaceous, 3–6 mm in diameter, and most commonly seen on the face (particularly in a periorbital distribution), the trunk, the extensor aspects of the extremities, and the neck (Figs 9.4, 9.5). In this variant the patient may also develop acute lymphadenopathy and uveitis, and a chest X-ray examination can reveal features of early respiratory involvement. Spontaneous resolution sometimes occurs. Occasionally, micropapular lesions are seen, particularly on the face and limbs (Fig. 9.6). Rarely patients develop sheets of pinhead-sized lichenoid papules on the trunk and limbs. The onset is abrupt and lesions may appear in crops. Some patients develop nodules and plaques, which may occur anywhere on the body, but most often affect the face, extremities, buttocks, and shoulders (Figs 9.7–9.11). Annular or serpiginous lesions are also encountered and sometimes there is a prominent telangiectatic component (angiolupoid sarcoid) (Figs 9.12, 9.13).10 Rarely, epidermal changes result in a psoriasiform or even ichthyosiform appearance.28 An exceptional case mimicking lipodermatosclerosis has been described.29 Chronic skin lesions are associated with pulmonary fibrosis, ocular, and bone involvement.
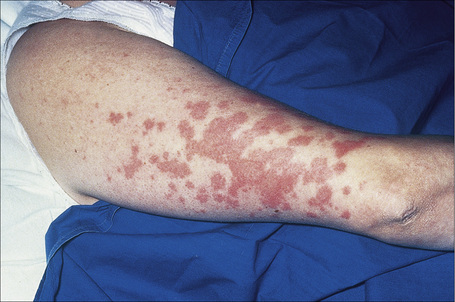
Fig. 9.4 Sarcoidosis: widespread erythematous plaques on the upper arm, some with an annular appearance.
By courtesy of R.A. Marsden, MD, St George’s Hospital, London, UK.
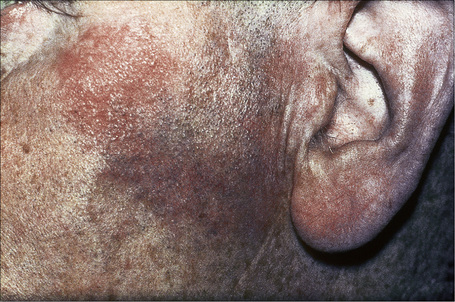
Fig. 9.5 Sarcoidosis: characteristic mauve plaque on the malar area with an infiltrative appearance.
By courtesy of R.A. Marsden, MD, St George’s Hospital, London, UK.
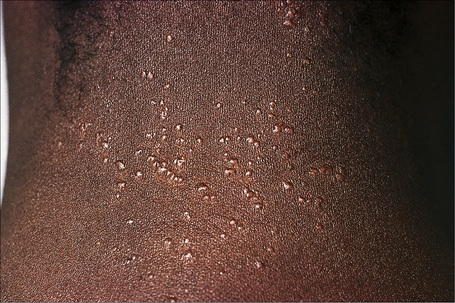
Fig. 9.6 Sarcoidosis: micropapular variant. Note the tiny lichenoid papules.
By courtesy of the Institute of Dermatology, London, UK.
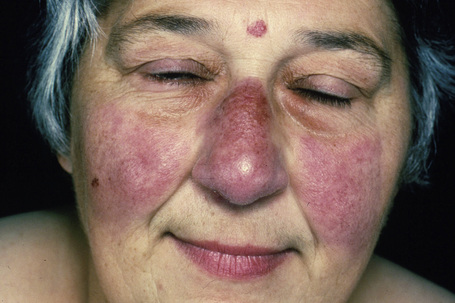
Fig. 9.7 Sarcoidosis: there is extensive facial involvement, a commonly affected site.
From the collection of the late N.P. Smith, MD, the Institute of Dermatology, London, UK.
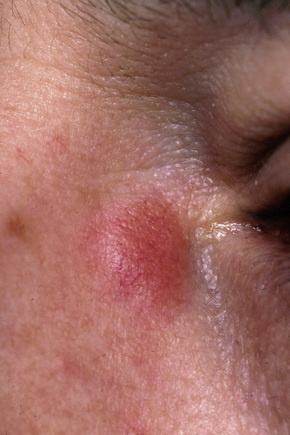
Fig. 9.8 Sarcoidosis: erythematous plaque adjacent to the eye.
From the collection of the late N.P. Smith, MD, the Institute of Dermatology, London, UK.
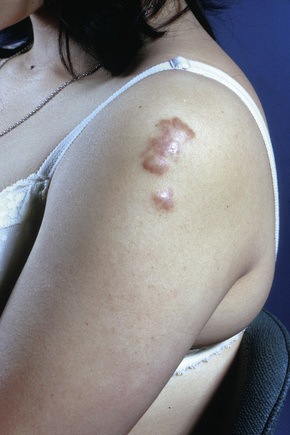
Fig. 9.9 Sarcoidosis: the extremities are commonly affected.
From the collection of the late N.P. Smith, MD, the Institute of Dermatology, London, UK.
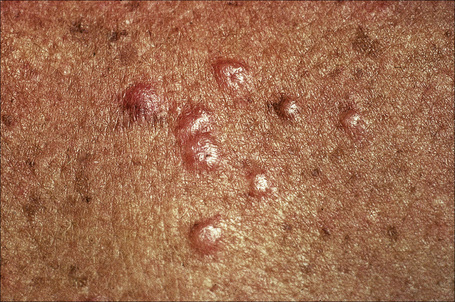
Fig. 9.10 Sarcoidosis: these grouped nodules are present on sun-damaged skin of the upper chest.
By courtesy of the Institute of Dermatology, London, UK.
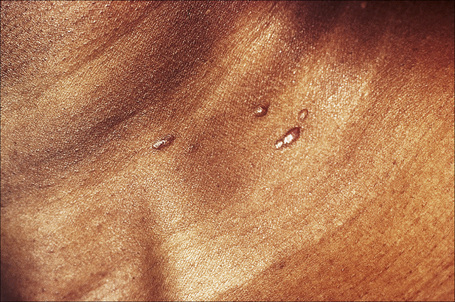
Fig. 9.11 Sarcoidosis: small nodules on the anterior aspect of the neck.
By courtesy of R.A. Marsden, MD, St George’s Hospital, London, UK.
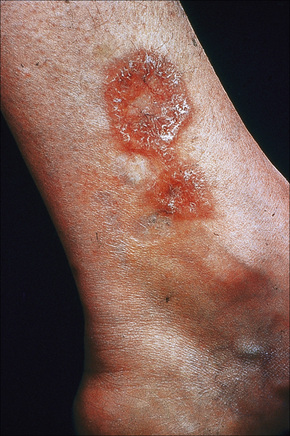
Fig. 9.12 Sarcoidosis: annular lesions on the ankle.
By courtesy of the Institute of Dermatology, London, UK.
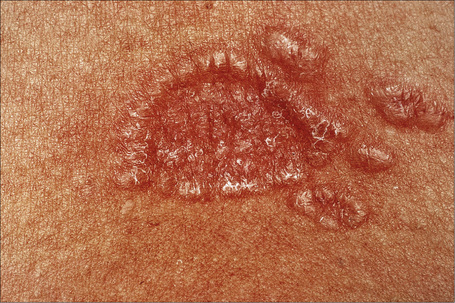
Fig. 9.13 Sarcoidosis: close-up view of an annular lesion. Note the beaded appearance.
By courtesy of the Institute of Dermatology, London, UK.
Most characteristic of sarcoidosis, however, is lupus pernio. This chronic violaceous plaque most often affects the nose, cheek, and ears, but lesions also sometimes affect the fingers and knees (Fig. 9.14). It is a particularly disfiguring variant and resolution is especially complicated by marked scarring. Lupus pernio is often associated with lesions in the upper respiratory tract and can be followed by nasal obstruction and septal perforation. Patients also have severe pulmonary fibrosis, bone cysts, and ocular lesions. This variant has an insidious onset and is associated with a prolonged course and poor prognosis.15
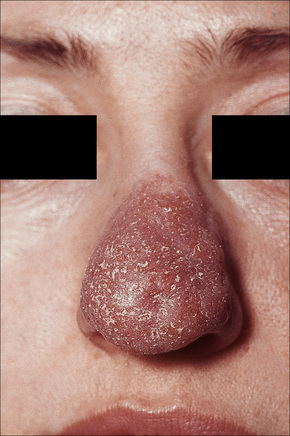
Fig. 9.14 Sarcoidosis: lupus pernio. The nose shows typical scaly violaceous swelling.
By courtesy of the Institute of Dermatology, London, UK.
Patients with sarcoidosis not uncommonly develop lesions in scar tissue30,31 and also in relation to trauma at the sites of desensitizing injections, tattoos, venipuncture, surgery, laser, cosmetic fillers, and BCG (Fig. 9.15).32–37 A single case attributed to copper-containing earrings has been described.38 Sarcoidal granulomata in association with foreign bodies do not necessarily imply a diagnosis of sarcoidosis. However, a small number of patients with sarcoidal granulomata in association with silica and tattoo pigment may have systemic sarcoidosis or the latter may develop subsequently.39 Awareness of this problem is important as such cutaneous granulomata may be the first manifestation of the disease. Sarcoidosis has also been documented presenting in a tattoo in association with interferon-alpha (IFN-α) treatment for chronic hepatitis C.40 Interestingly, sarcoidosis has also been reported rarely in patients receiving interferon and ribavirin for chronic hepatitis C,41,42 and in rare patients on interferon-alpha therapy for melanoma.43,44 Sarcoidosis in patients with chronic hepatitis C may present concomitantly with the disease and unrelated to medication, or more often triggered by treatment, mainly ribavirin and interferon-alpha.45

Fig. 9.15 Sarcoidosis: tattoo reaction. There are multiple dome-shaped nodules.
By courtesy of the Institute of Dermatology, London, UK.
Hypopigmented lesions may be seen in black patients.46 Unusual cutaneous manifestations include subcutaneous nodules, ichthyosiform lesions, erythroderma, scarring and nonscarring alopecia, lymphedema, nail dystrophy in the absence of underlying bone changes, verrucous lesions, generalized atrophy, leonine facies, palmar erythema, and leg ulcers with or without granulomatous vasculitis.47–61 Lesions of scarring alopecia may clinically mimic discoid lupus erythematosus.62 Subcutaneous nodules are rare and present as persistent, freely mobile, often painful lesions measuring 5–15 mm in diameter. It has been suggested that subcutaneous lesions are more often associated with systemic disease.63 In a further study the subcutaneous involvement occurred at the beginning of the disease and the associated systemic disease was not severe.64 Oral and genital involvement is rare but disease restricted to the vulva has been documented.65–67 Sarcoidosis has also been described presenting as a testicular mass.68
Ninety percent of patients with sarcoidosis have pulmonary involvement.27 Bilateral hilar lymphadenopathy is the commonest intrathoracic manifestation of sarcoidosis and together with pulmonary involvement forms the most frequent lesion. Intrathoracic manifestations in sarcoidosis are classified into five subgroups: 6,10
Systemic vasculitis involving small- to large-caliber vessels has been found in some adults and children with sarcoidosis.69 This manifestation tends to be more common in African-American and Asian patients.69
Neurological involvement occurs in 5–15% of patients with systemic sarcoidosis. Clinical manifestations include facial nerve palsy, Guillain-Barré syndrome, optic nerve disease, meningitis, seizure, and encephalopathy.6,70,71 In one study, neurosarcoidosis was the presenting symptom in 31% of patients.70 The combination of uveitis, facial nerve palsy, fever, and swelling of the parotid gland is known as uveoparotid fever (Heerfordt’s syndrome). This condition is often associated with central nervous system involvement. Hypothalamic and pituitary lesions are rare and may manifest as diabetes insipidus or panhypopituitarism.
Cardiac lesions are uncommon, but are of particular importance due to the associated mortality.6 In a recent autopsy series, 50% of all deaths were due to cardiac disease.72 This same study found that the clinician often does not appreciate the presence of cardiac involvement – the antemortem diagnosis of cardiac lesions was made in only 29% of patients.72 Granulomata may occur at any site, but appear to show a predilection for the conduction system. Clinical manifestations include ventricular tachycardia, complete heart block, congestive cardiac failure, pericardial effusion, and myocardial infarction. Sarcoidal granulomata may affect small and large blood vessels, in particular the pulmonary vasculature.
Although a rare complication, patients with sarcoidosis appear to be more prone to develop cryptococcosis than other infections.73
Radiologically demonstrable bone lesions occur in about 15% of patients. Early lesions consist of osteoporosis, cortical thinning, and mottled rarefaction. Established lesions are cystic and are sometimes associated with pathological fractures. The hands and feet are predominantly affected (Fig. 9.16). Destruction of the nasal bones can result from direct infiltration in patients with lupus pernio.
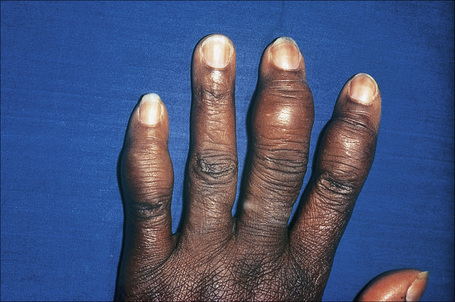
Fig. 9.16 Sarcoidosis: there is marked swelling of the distal interphalangeal joints.
By courtesy of R.A. Marsden, MD, St George’s Hospital, London, UK.
Hypercalcemia and, particularly, hypercalcuria are important complications of sarcoidosis. This is possibly due to increased intestinal absorption of calcium and abnormal production of 1,25-dihydroxyvitamin D.74 It is more often transitory, but in a small proportion of patients it is persistent and sometimes complicated by the development of renal failure due to nephrocalcinosis. Granulomata are found on histological examination of the kidney in up to 40% of patients.
The Kveim test was used in the past to aid in diagnosis. A homogenate of known sarcoid tissue is injected intradermally at a marked (India ink) site, and 4–6 weeks later the injection site is biopsied. A positive result depends upon the detection of an epithelioid cell granuloma. False-positive reactions may occur with other diseases including Crohn’s disease, infection (mycobacterial, fungal), berylliosis, silicosis, asbestosis, and lymphoma. The stimulatory ‘sarcoidal’ antigen of the Kveim reagent has not been identified.75 The Kveim test is almost never used nowadays because of difficulties in obtaining the homogenate of sarcoid tissue.
Although sarcoidosis is associated with a high morbidity, the mortality rate is low, being of the order of 3–6%. Causes of death include cardiac involvement and respiratory or renal failure. The prognosis is better in females and appears to be improved in those with a positive purified protein derivative (PPD) skin test and normal serum immunoglobulin levels. The severity of disease is greater in blacks and Asians compared with Caucasians.76 Of interest, despite the very marked upset in immunological phenomena, patients do not seem to have an associated greatly increased risk of opportunistic infections except as a consequence of therapy (e.g., corticosteroids).
The association between sarcoidosis and a number of systemic diseases is probably coincidental. Sarcoidosis has been documented in association with vitiligo, pernicious anemia, autoimmune thyroiditis, Graves’ disease, chronic hepatitis, Addison’s disease, Sjögren’s syndrome, diabetes mellitus and ulcerative colitis, lymphoma, human immunodeficiency virus (HIV) infection, and primary biliary chirrosis.77–88 Interestingly, patients with acquired immunodeficiency syndrome (AIDS) usually develop manifestations of sarcoidosis after antiretroviral therapy is started. This phenomenon is the result of the immune restoration syndrome.85,89 Associations with cutaneous autoimmune disease include dermatitis herpetiformis and linear IgA disease.90,91 A single case of trachyonychia associated with sarcoidosis has been reported.92
Pathogenesis and histological features
The pathogenesis of sarcoidosis is poorly understood. The demonstration of familial clustering suggests hereditary susceptibility to sarcoidosis in at least a subset of patients.14,93
Despite intensive studies, the etiology and pathogenesis of sarcoidosis remains elusive.94,95 It is likely, however, that sarcoidosis represents a reaction pattern that may develop in a predisposed patient on exposure to one or more infective agents or other antigens.
The role of mycobacteria in the pathogenesis of sarcoidosis is a controversial topic. Attempts at detection of mycobacterial DNA by polymerase chain reaction (PCR) have produced conflicting results. While some authors have failed to detect mycobacterial DNA, others have identified DNA of various strains of tuberculous and nontuberculous mycobacteria.96–100 In one study, although amplified mycobacterial DNA was detected by PCR in 38% of sarcoidosis patients, mycobacterial DNA was also detected in tissue in 44% of control patients.101 Furthermore, most studies published in the literature fail to report more than 6% positivity for Mycobacterium tuberculosis DNA in patients with sarcoidosis.102 In another interesting study, cell wall deficient acid-fast bacteria (L forms) were cultured from the blood of 19 of 20 patients with sarcoidosis but not from controls.103 In summary, these mixed results between laboratories have not clarified the role of mycobacteria in the pathogenesis of sarcoidosis. It seems, however, that mycobacteria may be of etiological importance in at least a subset of cases.
Propionibacterium acnes DNA has also been identified in tissues of patients with sarcoidosis, including involved lymph nodes.104,105 The significance of this finding remains uncertain. Human herpesvirus 8 DNA has not been demonstrated in tissues of patients with sarcoidosis.105
The occasional association with known autoimmune diseases, such as progressive systemic sclerosis and systemic lupus erythematosus (SLE), has inevitably led to the proposal of an autoimmune pathogenesis. Although many familial cases have been reported in the literature, no consistent pattern of inheritance has emerged. The results of human leukocyte antigen (HLA) typing have shown associations with particular features of the disease; for example, HLA-A1 and HLA-B8 are associated with arthritis, HLA-A1 is also associated with uveitis, and HLA-B13 may be associated with a chronic refractory variant.10 Patients with HLA-DR17 have a better prognosis.106 One study has shown that patients with sarcoidosis have an increased frequency of a glutamine residue at position 69 of the B1 chain of the HLA-DPB molecule compared with a control population.107 This is particularly interesting since a similar polymorphism has been documented in patients with chronic beryllium disease, a disorder also characterized by granulomata and which shares some pathological features in common with sarcoidosis.
Immunological investigations in patients with sarcoidosis have produced an immense wealth of data, which reveal that there is clearly an associated state of abnormal immunological hyperactivity. There are alterations of both cell-mediated and humoral immunity. Despite great efforts to clarify the immunobiology of sarcoidosis, particularly with regard to the precise antigens that may facilitate the disease, we still do not have a clear understanding of the disease process. Sarcoidosis, at least in part, appears to be due to a hyperactive T-helper cell proliferation with lymphokine production.108 Increased T-helper (Th1, Th2) cells are present in the alveolar lung parenchyma. Several studies have demonstrated selective activation of certain oligoclonal T-cell subsets.109–112 In one study, there was a correlation between the particular oligoclonal T-cell subsets and disease activity.109 Th1 lymphocytes (T cells expressing interleukin (IL)-2 and IFN-γ) preferentially accumulate in pulmonary parenchyma and the alveolar space compared with Th2 lymphocytes (T cells expressing IL-4 and IL-5).113 Compared with T-cells in peripheral blood, T cells obtained by bronchoalveolar lavage show greater expression of IFN-γ and tumor necrosis factor alpha (TNF-α).106 Of interest, patients with HLA-DR17 show a muted cytokine response, a finding that is perhaps related to the better prognosis observed in this subset of patients.107
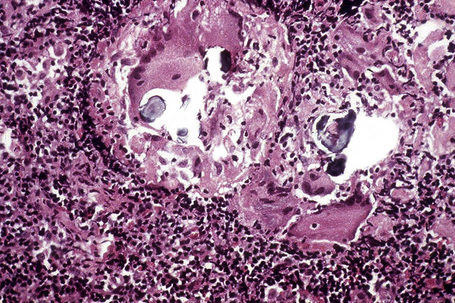
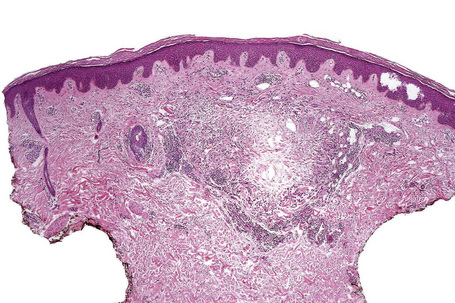
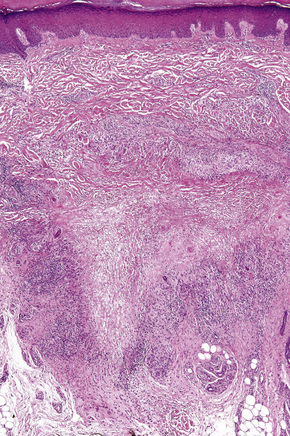
Histologically, sarcoidosis is characterized by a dense, noncaseating granulomatous infiltrate in the dermis (Figs. 9.17, 9.18), which sometimes extends into the subcutaneous fat. The granulomata are discrete and strikingly uniform in size and shape. They are composed of epithelioid histiocytes with abundant eosinophilic cytoplasm and oval or twisted vesicular nuclei often containing a small central nucleolus (Fig. 9.19). Variable numbers of Langhans giant cells are present and sometimes a scattering of lymphocytes is seen at the peripheral margin of the granuloma (Fig. 9.20). Discrete small central foci of fibrinoid necrosis are sometimes present but caseation necrosis is rare (Fig. 9.21).114,115 Transepidermal elimination is sometimes seen.116 The epidermis is usually normal although occasional cases display acanthosis and sometimes the granulomata are focally lichenoid. A predominantly lichenoid pattern may exceptionally be seen.117 Exceptional cases of sarcoidosis may display histologic findings that focally overlap with granuloma annulare, palisading neutrophilic and granulomatous dermatitis, and interstitial granulomatous dermatitis.118 Further histologic findings described include elastophagocytosis, perineural granulomas resembling leprosy, mucin deposition, and an infiltrate rich in plasma cells.115
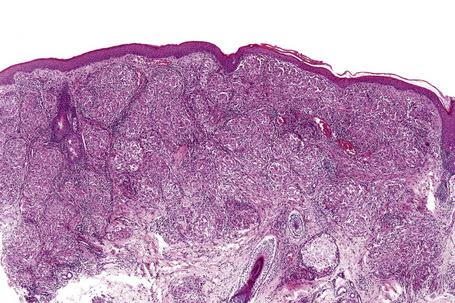
Fig. 9.17 Sarcoidosis: the dermis is replaced by uniform circumscribed nests of non-caseating granulomata.
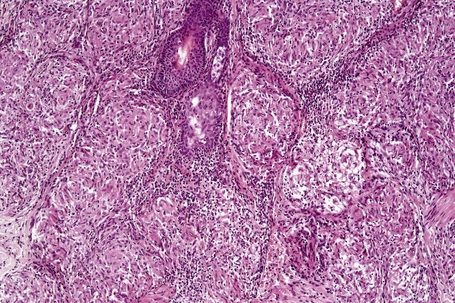
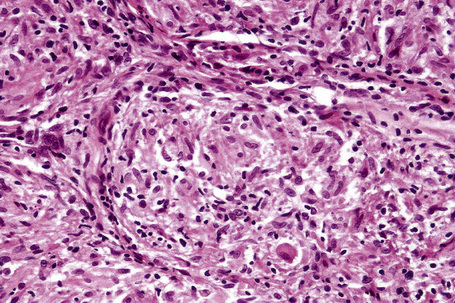
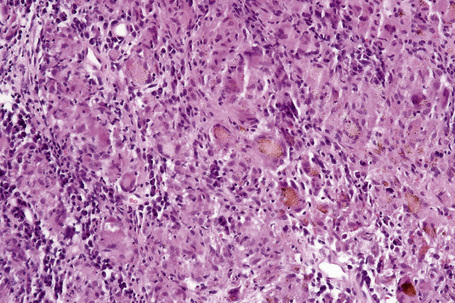
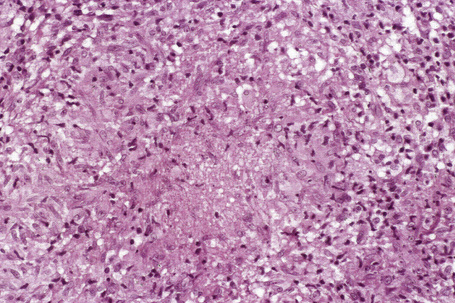
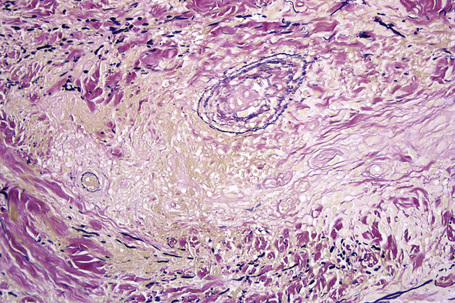
In some cutaneous lesions, inclusion bodies are present, although much less frequently than in lymph nodes. The Schaumann body, a basophilic, laminated, rounded, conchoidal structure composed of calcium carbonate, calcium oxalate, phosphate, iron, and dolomite, is not specific for sarcoidosis and is seen in a number of other granulomatous conditions including tuberculosis and berylliosis (Fig. 9.22).119–121 The asteroid body is a small intracytoplasmic eosinophilic star-shaped structure; it is not specific for sarcoidosis, being seen also, for example, in tuberculosis, tuberculoid leprosy, berylliosis, and atypical facial necrobiosis. It is also commonly found in necrobiotic xanthogranuloma.122 Initial studies suggested that the asteroid body was composed of collagen but more recent reports, using immunohistochemistry, suggest that it is a product of the microtubular system.123,124 The presence of foreign material in sarcoidal granulomata does not exclude the diagnosis of sarcoidosis. In fact, polarizable material has been found in up to 5% of cases.125–127
It has been shown that the gli-1 oncogene is consistently and abnormally expressed in the cells forming the granulomata not only in sarcoidosis but also in granuloma annulare and necrobiosis lipoidica. This observation raises the possibility of trials using inhibitors of gli-1 signaling to treat this group of granulomatous disorders.128
Differential diagnosis
Sarcoidosis must be approached as a diagnosis of exclusion and has to be distinguished from the numerous conditions that may be associated with a noncaseating granulomatous histology, including some forms of tuberculosis, tuberculoid leprosy, berylliosis, fungal infections, Crohn’s disease, and foreign body granulomatous reactions.129 Therefore, the use of special stains, including the Ziehl-Neelsen preparation for mycobacteria and the periodic acid-Schiff (PAS) and methenamine silver reactions for fungi, is mandatory before diagnosing sarcoidosis. Depending on the clinical context, culture may also be required to exclude an infective etiology. Tuberculoid leprosy is characterized by nerve involvement, a feature that is usually absent in sarcoidosis.
Some of the granulomata seen in a variety of primary immunodeficiency syndromes closely mimic those found in sarcoidosis and histological distinction may be impossible. A study comparing granulomata in sarcoidosis to those seen in primary immunodefiencies found a much lower rate of CD4+/CD8+ cells in the former as opposed to the latter.130
Labial and gingival involvement may be histologically mistaken for Crohn’s disease and granulomatous cheilitis (Miescher). It is worth noting that in rare cases oral involvement in Crohn’s disease may precede systemic manifestations by several years. Metastatic Crohn’s disease may be difficult to distinguish from sarcoidosis. The former often show nonsuppurative granulomata in a diffuse pattern and surrounded by a thin cuff of lymphocytes. Further frequent findings include the presence of numerous eosinophils and ulceration, findings not often seen in sarcoidosis.131
Granulomatous lesions that have been described in exogenous ochronosis appear to be related to sarcoidosis.132 However, similar lesions have also been described as showing changes mimicking actinic granuloma.133
Granuloma annulare
Clinical features
Granuloma annulare is a common, usually asymptomatic, dermatosis of unknown etiology.1,2 It may be divided into six clinical subsets:
Unusual clinical variants include pustular follicular lesions and presentation with patches.3,4 A single case presenting as contact dermatitis has been reported.5 Granuloma annulare (often with widespread disseminated lesions) has been described in patients with HIV infection and sometimes may be the presenting sign.6–18 Granuloma annulare, mainly the generalized variant (see below), has also been reported in association with both Hodgkin’s and non-Hodgkin’s lymphoma.19–22 Exceptionally, anterior uveitis and concomitant skin lesions have been described.23,24
Other documented associations of granuloma annulare include morphea, chronic hepatitis C infection, autoimmune thyroiditis, secondary hyperparathyroidism, sarcoidosis, Plummer’s disease, myelodysplastic syndrome, metastatic carcinoma, and a bee sting.25–33 Granuloma annulare has also been described after vaccination for tetanus and diphtheria, hepatitis B and tuberculosis (BCG), and after mesotherapy.34–38 It may also develop in the scars of herpes zoster.39–42 It is important to highlight that most patients with the condition heal after variable periods of time, and long follow-up has not revealed consistent associations with any systemic diseases.43 A further study has found no consistent relationship between malignant neoplasms and granuloma annulare.44 However, it has been suggested that elderly patients with lesions that do not have typical features of granuloma annulare but display microscopic findings resembling granuloma annulare should be investigated for an underlying malignancy, especially lymphoma.44
Granuloma annulare has developed during treatment with allopurinol, amlodipine, daclizumab, and antitumor necrosis factor.45–48 Interferon-alpha has been associated with generalized interstitial granuloma annulare.49 It is most likely, however, that granuloma annulare-like eruptions secondary to drug administration often represent interstitial granulomatous drug eruptions.
Localized granuloma annulare
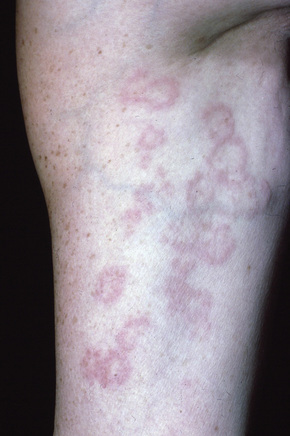
The localized variant is the commonest type. It usually presents in the first three decades and is associated with a female preponderance (2.25:1). Lesions consist of one or several papules, which may be skin-colored, red or violaceous, and are typically distributed to form an annular or arcuate lesion 1–5 cm in diameter (Figs 9.23–9.27). About 50% of patients have solitary lesions. The acral sites are most commonly affected, in particular the knuckles and dorsum of the fingers. In a small proportion of patients, lesions are present on both the upper and lower limbs, and occasionally the trunk is affected. Lesions on the palms are exceptional.50 Facial involvement appears to be uncommon.51,52 In a reported case, lesions were restricted to the area involved by a Becker’s nevus.53 Although lesions may be persistent, approximately 50% of patients can anticipate resolution by about 2 years from onset. However, recurrences are, unfortunately, quite common. Patients in which the disease arises earlier in life appear to have earlier resolution of lesions.54 Interestingly, on occasion lesions regress spontaneously after biopsy.55 In one case, spontaneous resolution resulted in mid-dermal elastolysis.56 Rarely, granuloma annulare has been reported in families and in monozygotic twins.57 A case has been documented in which the lesions recurred seasonally with sun-exposed areas.58 There has only been a single case report of cutaneous granuloma annulare with similar lesions in an intra-abdominal location.59 In one case, granuloma annulare was the first sign of adult T-cell leukemia/lymphoma and in a further patient it was associated with angioimmunoblastic T-cell lymphoma.60,61 Very rarely, acral, localized granuloma annulare may present as an acute and painful eruption.62
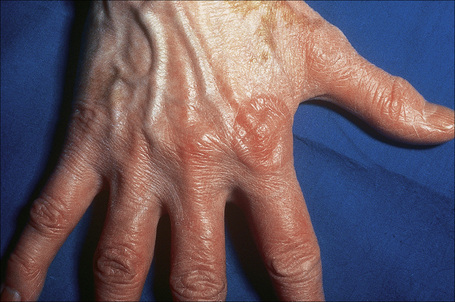
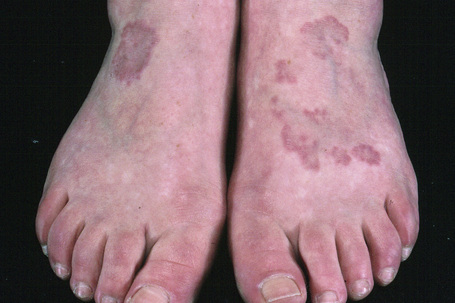
Fig. 9.24 Localized granuloma annulare: in this patient multiple lesions are present on the feet.
From the collection of the late N.P. Smith, MD, the Institute of Dermatology, London, UK.
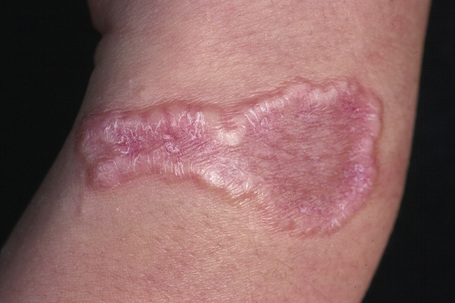
Fig. 9.25 Localized granuloma annulare: this arm lesion shows a characteristic beaded margin.
From the collection of the late N.P. Smith, MD, the Institute of Dermatology, London, UK.
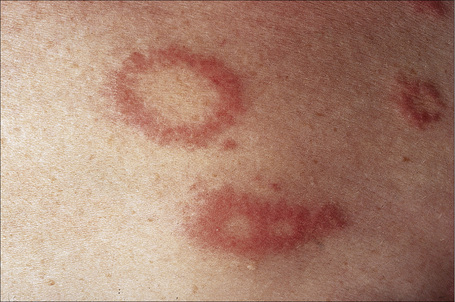
Fig. 9.26 Localized granuloma annulare: close-up view of annular lesions.
By courtesy of the Institute of Dermatology, London, UK.
Generalized granuloma annulare
Generalized lesions occur in approximately 15% of patients with granuloma annulare.2,63 As with the localized form, there is an increased incidence in females; however, the median age differs the majority of cases occurring in patients in the fourth to seventh decades, with the rest appearing during the first decade. Patients with generalized granuloma annulare have an increased incidence of HLA-Bw35.64 Generalized granuloma annulare is defined as lesions occurring on at least the trunk and either upper or lower extremities, or both.2 Most lesions are papules, which may be distributed in an annular pattern, but maculopapules and nodules also occur. They vary in hue from flesh-colored or red, to tan, brown or yellow. Numbers vary from several dozen to hundreds (Figs 9.28 to 9.30). A single patient has been documented with generalized disease accompanied by marked swelling of the hands and another patient developed the disease following erythema multiforme.65,66 Lesions may be asymptomatic or pruritic.2 As with the localized form, the disease is persistent, but some patients experience resolution within 4 years. Anetoderma has been exceptionally reported as a complication of generalized granuloma annulare.67 A remarkable association with giant cell arteritis, gastrointestinal stromal tumor, and other internal malignancies including ovarian and gastric cancer has been reported.68–70 Tuberculous lymphadenits was an association in one case.71 In two instances, the condition was the initial manifestation of chronic myelomonocytic leukemia.72 An association with lymphoma, including Hodgkin’s disease, has also been described.73 A patient with hepatitis B developed generalized granuloma annulare, and viral DNA was detected in the skin lesions by PCR.74 A further case presented in a photosensitive distribution and healed with scarring and milia formation.75
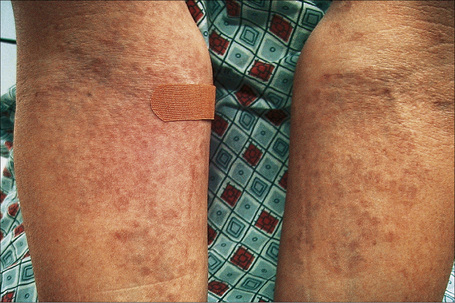
Fig. 9.28 Generalized granuloma annulare: innumerable papules are present on this patient’s arms.
By courtesy of J. Williams, MD, Brigham and Women’s Hospital, Boston, USA.
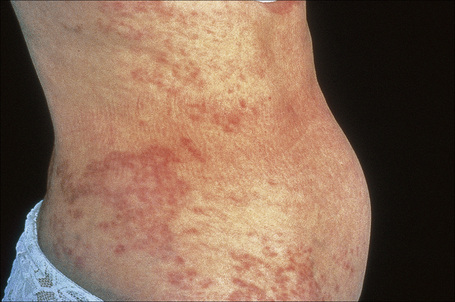
Fig. 9.29 Generalized granuloma annulare: there are widespread papules and plaques.
By courtesy of the Institute of Dermatology, London, UK.
Perforating granuloma annulare
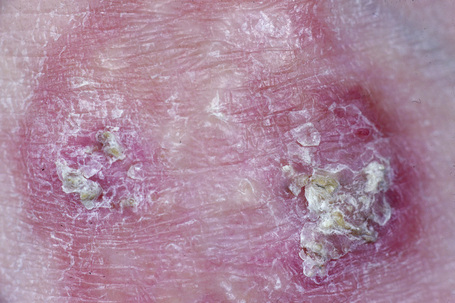
Perforating granuloma annulare is distinguished by the presence of transepidermal elimination of necrobiotic collagen.10,18,76–79 Clinically, the lesion presents as a group of papules with an umbilicated crust usually located on the extremities, often the dorsum of the hands (Fig. 9.31). Presentation of lesions on the ears has exceptionally been described, as has a generalized variant.80–82 It may affect both children and adults, and both localized and generalized forms exist. Spontaneous resolution sometimes occurs within months or years of onset. An exceptional case of perforating granuloma annulare which developed following tattooing has been reported.83
Subcutaneous (deep) granuloma annulare
The subcutaneous variant is synonymous with the pseudorheumatoid nodule of childhood and deep granuloma annulare.84–87 Lesions may present de novo or arise in association with typical cutaneous papules. It occurs in childhood, often affecting the underlying periosteum and involving predominantly the lower legs (particularly the tibia), feet, buttocks, hands, and head.88 Lesions may also present on the penis or eyelid.89,90 An exceptional case of numerous lesions limited to the scalp of a child which regressed spontaneously has been reported.91 A further patient presented with a periorbital subperiostal lesion and in another patient the lesion was congenital.92,93 In one study of 47 patients, the mean age was 4.3 years.88 In some instances, there is a past history of trauma. By definition, such children do not have rheumatoid arthritis or rheumatic fever. The lesion usually regresses after several years. However, recurrences appear in 19% of patients.88
Papular granuloma annulare
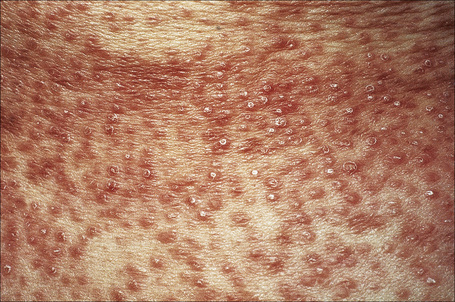
Papular granuloma annulare presents as flesh-colored or hypopigmented, 1–3-mm diameter papules on the dorsal aspect of the hands, usually in male children. Occasional lesions may be umbilicated or generalized (Figs 9.32, 9.33).94
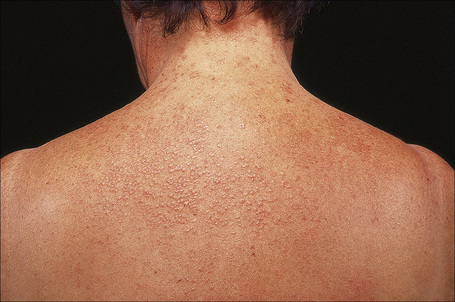
Fig. 9.32 Papular granuloma annulare: widespread papules are present on this patient’s back and shoulders.
By courtesy of the Institute of Dermatology, London, UK.
Pathogenesis and histological features
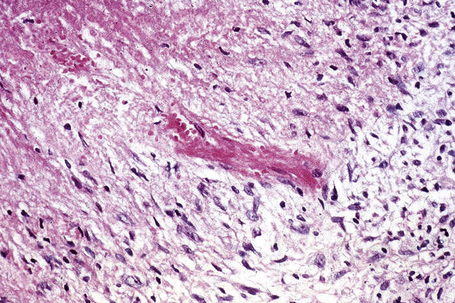
The cause of granuloma annulare is unknown. The original concept that it represented a tuberculid has long since been discounted. Although it has been reported at the site of previous herpes zoster infection and verruca vulgaris, it is unlikely that an infectious pathogenesis exists. Borrelia has been demonstrated by focus-floating microscopy in a number of biopsies of patients with granuloma annulare raising the possibility of a pathogenetic role for the organism in some cases of the disease.98 However, a study based on PCR found no association between granuloma annulare and Borrelia infection.99 There is a wide variety of currently possible pathogenetic mechanisms, most of which have some merit, but none of which satisfactorily clarifies the precise mechanism by which the lesions of granuloma annulare develop.100 Particularly popular are an immune complex vasculitic process and a cell-mediated delayed hypersensitivity reaction. Evidence in favor of the former has been the detection, by direct immunofluorescence, of immunoreactants (IgM and complement) in blood vessel walls in some patients.100 Elevated levels of circulating immune complexes have also been recorded.101 The histology may reveal features suggestive of a vasculitic process, including endothelial swelling, vessel wall thickening (due to the deposition of PAS-positive material), vascular occlusion, and necrosis (Fig. 9.34).102 All of the latter changes may, of course, develop as a consequence of the inflammatory process rather than cause it. A recent study of serial sections of 38 biopsies in 35 patients found no evidence of a vasculitic process in any of the cases.103
In favor of a cell-mediated delayed hypersensitivity reaction are:
It has been suggested that Th1 lymphocytes expressing interferon-gamma induce a delayed hypersensitivity reaction leading to macrophages becoming aggressive effector cells that express tumor necrosis factor-alpha and matrix metalloproteinases.105 If tumor necrosis factor alpha plays a role in the induction of the disease, then agents that block this cytokine may be useful in treating the condition. Although some patients respond to these agents other do not and the reason for this is not clear. Monozygotic twins with generalized granuloma annulare and the 8.1 ancestral haplotype, a genotype that leads to increased production of tumor necrosis alpha, have responded well to adalimumab.106
Patients with granuloma annulare may have raised serum migration inhibition factor activity.87 Defective neutrophil migration has also been reported.107,108 Other proposed pathogenetic mechanisms include collagen damage by macrophage lysosomal hydrolytic enzymes as the initial event, or a primary disorder of collagen leading to an allergic or nonallergic tissue reaction. The increased incidences of diabetes mellitus and HLA-B8 may also be of pathogenetic significance (compare with necrobiosis lipoidica).109 In a study of a group of pediatric patients with multiple lesions of granuloma annulare it was found that they had significantly lower serum insulin values than the control group and showed mild impairment of glucose tolerance.110 However, these children, often had a family history of diabetes mellitus.
Although there are reports of generalized granuloma being associated with sunlight, this appears to be of doubtful significance.2
It has been shown that the glioma-associated oncogene homologue gli-1, a member of the vertebrate zinc finger transcription factor genes of the gli superfamily is highly expressed in a number of granulomatous disorders including granuloma annulare.111 The relevance of this in the pathogenesis of granuloma annulare is not clear but it raises the possibility of using inhibitors of gli-1 signaling in the treatment of granulomatous noninfectious diseases.
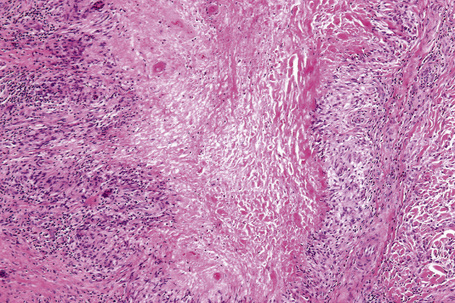
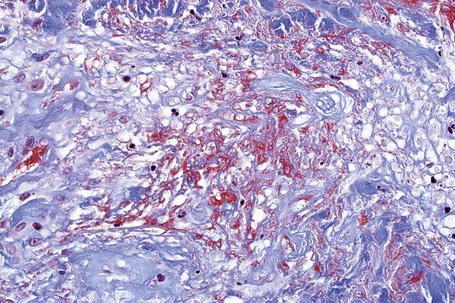
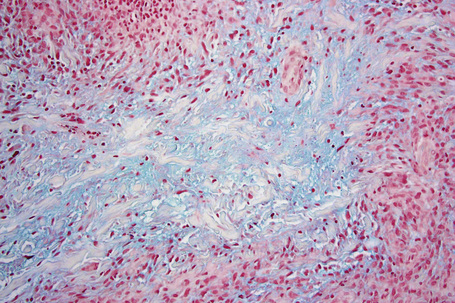
The most characteristic histological lesion seen in granuloma annulare is the palisading granuloma (Figs 9.35–9.38). This consists of a central core of degenerate (necrobiotic) collagen, surrounded by an often radially arranged infiltrate of lymphocytes, histiocytes, and fibroblasts. Elastic tissue may be absent within these foci and there can be phagocytosis of elastic fibers by giant cells at the periphery of the granuloma (Fig. 9.39).112 However, altered elastic fibers are not a constant finding. Solar elastosis is not a feature of granuloma annulare. In some lesions the altered collagen has a somewhat basophilic appearance due to the presence of acid mucopolysaccharides, but more commonly there is eosinophilia, due in part to fibrin deposition (Fig. 9.40). Heparin sulfate is an important component of the mucin in granuloma annulare but not of other cutaneous diseases associated with mucin deposition (Fig. 9.41).113
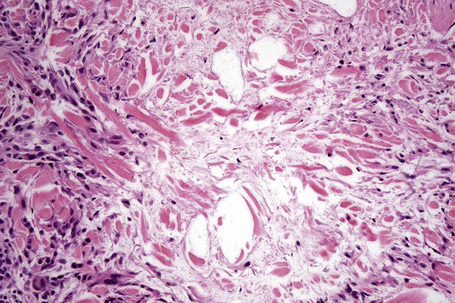
Occasionally, sparse karyorrhectic debris is present in the center of the lesion and sometimes the necrobiotic foci contain lipid droplets. More often, however, the collagenous degeneration is not organized into a nodular pattern, but affects isolated fibers in a random pattern, an appearance often best appreciated on low-power examination (Fig. 9.42).114 In this so-called diffuse or interstitial form of granuloma annulare, affected fibers, which are swollen and intensely eosinophilic, alternate with apparently normal fibers to give a rather disorganized appearance (Figs. 9.43, 9.44). Necrobiosis is minimal or absent. Characteristically, the collagen fibers are separated by mucin, which stains positively with Alcian blue at pH 2.5. Histiocytes are often seen infiltrating around and between affected fibers, and this feature may be a helpful clue to the diagnosis in early cases when the collagen changes are inconspicuous and should, therefore, encourage examination of additional sections to detect more typical features (Fig. 9.45).
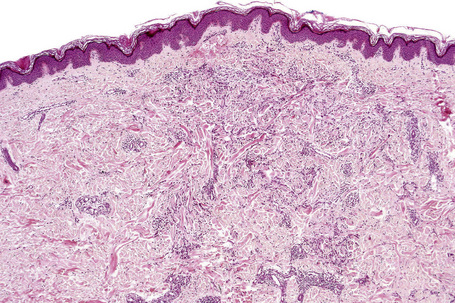
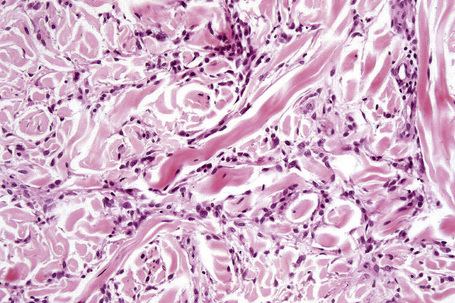
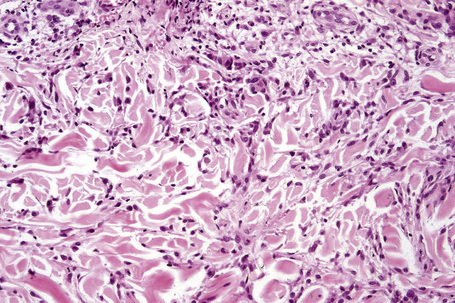
Fig. 9.44 Diffuse granuloma annulare: higher-power view showing the dense interstitial histiocytic infiltrate.
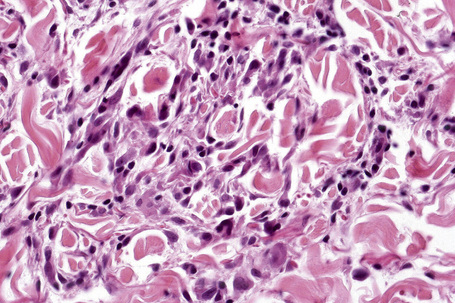
An almost inevitable feature of granuloma annulare is the presence of a perivascular chronic inflammatory cell infiltrate, both within the lesion and in the adjacent tissue. Well-formed sarcoidal granulomata with associated giant cells are seen in some cases. Significant numbers of eosinophils may be encountered.115 In one study, eosinophils were present in 66% of biopsies, of which 14% showed more than 10 eosinophils per high-power field.115 Plasma cells are rare and this is useful in the differential diagnosis with necrobiosis lipoidica (see below). Neutrophils are a rare finding and when present, particularly in association with changes of vasculitis, it is likely that there is an association with systemic disease.116
Although the relationship with Borrelia infection is debatable, it has been suggested that formation of pseudorosettes in granuloma annulare may suggest infection with the organism.117
In perforating granuloma annulare the necrobiotic debris is present in close proximity to the epidermis and may be seen to be engulfed by the latter to form a perforating channel by which the necrotic material is extruded to the surface (Figs. 9.46, 9.47). If serial sections are performed, the perforation is often shown to occur through a hair follicle.
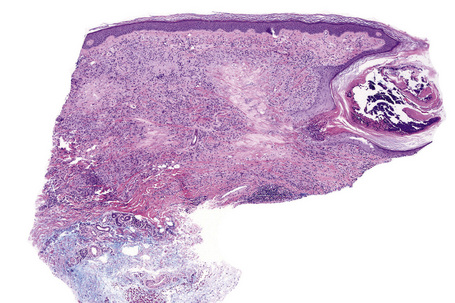
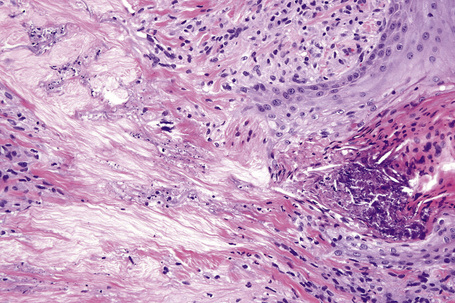
The subcutaneous lesions are much larger than the superficial ones (Figs. 9.48, 9.49) and are frequently composed of multiple nodules. There is usually massive necrobiosis and abundant mucin; on occasions, lipid droplets are evident. Mucin, however, may be minimal or not apparent and if fibrin deposition is present, distinction from rheumatoid nodule is impossible. A dense rim of lymphocytes, histiocytes, and fibroblasts surrounds the necrobiotic center. Multinucleate giant cells are common and eosinophils are often present. The latter appear to be more common in this variant than in ordinary granuloma annulare. Fibrosis of the surrounding tissue may be marked. In up to 25% of cases of subcutaneous granuloma annulare, changes of classic granuloma annulare may be seen in the dermis.118
< div class='tao-gold-member'>
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


