Gout and Hyperuricemia
KEY CONCEPTS
![]() In the absence of a history of gout, asymptomatic hyperuricemia may not require treatment.
In the absence of a history of gout, asymptomatic hyperuricemia may not require treatment.
![]() Acute gouty arthritis may be treated effectively with short courses of high-dose nonsteroidal antiinflammatory drugs (NSAIDs), corticosteroids, or colchicine.
Acute gouty arthritis may be treated effectively with short courses of high-dose nonsteroidal antiinflammatory drugs (NSAIDs), corticosteroids, or colchicine.
![]() Low-dose colchicine is highly effective at relieving acute attacks of gout; dose titration leads to more adverse effects but does not improve efficacy.
Low-dose colchicine is highly effective at relieving acute attacks of gout; dose titration leads to more adverse effects but does not improve efficacy.
![]() Treatment with urate-lowering drugs to reduce risk of recurrent attacks of gouty arthritis is considered cost-effective for patients having two or more attacks of gout per year.
Treatment with urate-lowering drugs to reduce risk of recurrent attacks of gouty arthritis is considered cost-effective for patients having two or more attacks of gout per year.
![]() Xanthine oxidase inhibitors are efficacious for the prophylaxis of recurrent gout attacks in both underexcreters and overproducers of uric acid. Either allopurinol or febuxostat should be initiated in patients with one of the following indications for urate-lowering therapy: (a) two or more gout attacks per year, (b) the presence of one or more tophus, (c) chronic kidney disease (stage 2 or worse), or (d) a history of urolithiasis. The dose of the xanthine oxidase inhibitor should be titrated to a goal serum urate concentration of <6 mg/dL (or <5 mg/dL if signs of gout persist at a level of 6 mg/dL).
Xanthine oxidase inhibitors are efficacious for the prophylaxis of recurrent gout attacks in both underexcreters and overproducers of uric acid. Either allopurinol or febuxostat should be initiated in patients with one of the following indications for urate-lowering therapy: (a) two or more gout attacks per year, (b) the presence of one or more tophus, (c) chronic kidney disease (stage 2 or worse), or (d) a history of urolithiasis. The dose of the xanthine oxidase inhibitor should be titrated to a goal serum urate concentration of <6 mg/dL (or <5 mg/dL if signs of gout persist at a level of 6 mg/dL).
![]() Uricosuric agents should be avoided for patients with renal impairment [a creatinine clearance below 50 mL/min (0.84 mL/s)], a history of renal calculi, or overproduction of uric acid.
Uricosuric agents should be avoided for patients with renal impairment [a creatinine clearance below 50 mL/min (0.84 mL/s)], a history of renal calculi, or overproduction of uric acid.
![]() Low-dose colchicine, NSAID, or corticosteroid therapy should be administered during the first 3 to 6 months of urate-lowering therapy to minimize the risk of acute gout attacks that may occur during this initiation period.
Low-dose colchicine, NSAID, or corticosteroid therapy should be administered during the first 3 to 6 months of urate-lowering therapy to minimize the risk of acute gout attacks that may occur during this initiation period.
![]() Uric acid nephrolithiasis should be treated with adequate hydration (2 to 3 L/day), a daytime urine-alkalinizing agent, and 60 to 80 mEq/day (60 to 80 mmol/L) of potassium bicarbonate or potassium citrate.
Uric acid nephrolithiasis should be treated with adequate hydration (2 to 3 L/day), a daytime urine-alkalinizing agent, and 60 to 80 mEq/day (60 to 80 mmol/L) of potassium bicarbonate or potassium citrate.
![]() Patients with hyperuricemia or gout should undergo comprehensive evaluation for signs and symptoms of cardiovascular disease, and aggressive management of cardiovascular risk factors (i.e., weight loss, reduction of alcohol intake, control of blood pressure, glucose, and lipids) should be undertaken as indicated.
Patients with hyperuricemia or gout should undergo comprehensive evaluation for signs and symptoms of cardiovascular disease, and aggressive management of cardiovascular risk factors (i.e., weight loss, reduction of alcohol intake, control of blood pressure, glucose, and lipids) should be undertaken as indicated.
The term gout describes a heterogeneous clinical spectrum of diseases including elevated serum urate concentration (hyperuricemia), recurrent attacks of acute arthritis associated with monosodium urate crystals in synovial fluid leukocytes, deposits of monosodium urate crystals (tophi) in tissues in and around joints, interstitial renal disease, and uric acid nephrolithiasis.1
The underlying metabolic disorder of gout is hyperuricemia, defined physiochemically as serum that is supersaturated with monosodium urate. At 37°C (98.6°F), serum urate concentrations above (or around) 7 mg/dL (416 μmol/L) begin to exceed the limit of solubility for monosodium urate.1 For determination of the risk of gout, hyperuricemia is defined statistically as serum urate concentrations greater than two standard deviations above the population means for age- and sex-matched healthy populations, usually 7 mg/dL (416 μmol/L) for men and 6 mg/dL (357 μmol/L) for women.1,2 Although hyperuricemia is fundamental to the development of gout, the mere presence of hyperuricemia itself is often an asymptomatic condition.
EPIDEMIOLOGY
Historically, gout has been referred to as the “disease of kings” since it was often associated with affluent societies and lifestyles of overindulgence, gluttony, and intemperance.1 Gout continues to occur more commonly in developed countries (e.g., United States, Japan, United Kingdom, and Australia) as compared to developing countries (e.g., China).3 In the United States, the prevalence of gout is increasing. According to data from the 2007 to 2008 National Health and Nutrition Examination Survey (NHANES), the prevalence of gout in US adults is 3.9%, which corresponds to an estimated 8.3 million people. This represents a 1.2% increase in prevalence compared with NHANES-III survey data from 1988 to 1994.4
Elevated serum urate levels are the single most important risk factor for the development of gout, and the relationship between the risk of an attack of acute gouty arthritis and serum urate levels is linearly correlated. The 5-year cumulative risk of gout for patients with serum urate concentrations <7 mg/dL (<416 μmol/L) is 0.6%, compared with a risk of 30.5% for those with urate levels >10 mg/dL (>595 μmol/L).5 Sustained elevation of serum urate is virtually essential for the development of gout; however, hyperuricemia does not always lead to gout, and many patients with hyperuricemia remain asymptomatic.2 Although unusual, acute gouty arthritis has been reported to occur in the presence of normal serum uric acid concentrations.6 The prevalence of hyperuricemia in the United States mirrors the trend seen with gout, affecting 21.4% of adults (43.3 million people) in 2007 to 2008 compared to just 18.2% in 1998 to 1994.4
The increased prevalence of gout and hyperuricemia may be partly explained by the aging of the population. Gout and hyperuricemia occur more commonly in the older adult with the highest prevalence, 12.6%, in those 80 years of age and older compared with just 0.4% in those ages 20 to 29 years.4 Another major contributor to the increased prevalence of gout in the United States is the obesity epidemic. Obese persons are twice as likely to have gout as nonobese counterparts.7 Dietary and lifestyle factors linked to obesity have also been independently associated with gout. These include consumption of alcohol, sugary beverages, and red meat along with a sedentary lifestyle.8
Regarding sex distribution, gout affects men about three times more often than women.4 The lowest rates of gout are observed in women younger than 45 years, approximately 0.6 cases per 1,000 person-years.9 Serum uric acid levels in women approach those of men once menopause has occurred; thus, in older age groups the gender gap narrows, and approximately half of newly diagnosed cases of gout are found in women.10,11 Gout in men younger than 30 years of age or in premenopausal women may indicate an inherited enzyme defect or the presence of renal disease. Although no genetic marker has been isolated for gout, the familial nature of gout strongly suggests an interaction between genetic and environmental factors.
ETIOLOGY AND PATHOPHYSIOLOGY
In humans, the production of uric acid is the terminal step in the degradation of purines. Uric acid serves no known physiologic purpose and is regarded as a waste product. Normal uric acid levels are near the limits of urate solubility, because of the delicate balance that exists between the amount of urate produced and excreted.2 Humans have higher uric acid levels than other mammals because they do not express the enzyme uricase, which converts uric acid into the more soluble allantoin.10
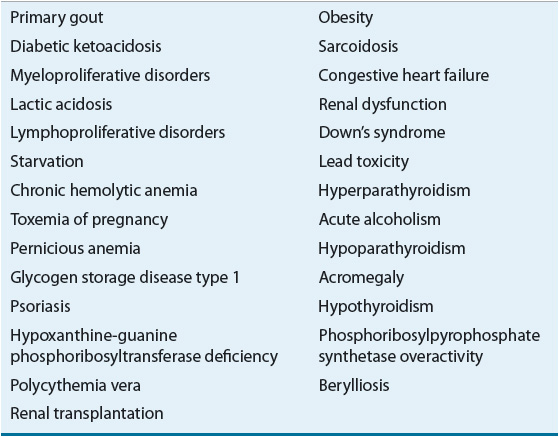
Gout occurs exclusively in humans in whom a miscible pool of uric acid exists. Under normal conditions, the amount of accumulated uric acid is about 1,200 mg in men and about 600 mg in women. The size of the urate pool is increased severalfold in individuals with gout. This excess accumulation may result from either overproduction or underexcretion of uric acid. Several conditions are associated with either decreased renal clearance or an overproduction of uric acid, leading to hyperuricemia. Table 74–1 lists some of these conditions.
TABLE 74-1 Conditions Associated with Hyperuricemia
Overproduction of Uric Acid
The purines from which uric acid is produced originate from three sources: dietary purine, conversion of tissue nucleic acid into purine nucleotides, and de novo synthesis of purine bases. The purines derived from these three sources enter a common metabolic pathway leading to the production of either nucleic acid or uric acid. Under normal circumstances, uric acid may accumulate excessively if production exceeds excretion. The average human produces about 600 to 800 mg of uric acid each day. Dietary purines play an unimportant role in the generation of hyperuricemia in the absence of some derangement in purine metabolism or elimination. However, diet modifications are important for patients with such problems who develop symptomatic hyperuricemia.
Several enzyme systems regulate purine metabolism. Abnormalities in these regulatory systems can result in overproduction of uric acid. Uric acid may also be overproduced as a consequence of increased breakdown of tissue nucleic acids and excessive rates of cell turnover, as observed with myeloproliferative and lymphoproliferative disorders, polycythemia vera, psoriasis, and some types of anemias. Cytotoxic medications used to treat these disorders can result in overproduction of uric acid secondary to lysis and breakdown of cellular matter.
Two enzyme abnormalities resulting in an overproduction of uric acid have been well described (Fig. 74–1). The first is an increase in the activity of phosphoribosyl pyrophosphate (PRPP) synthetase, which leads to an increased concentration of PRPP. PRPP is a key determinant of purine synthesis and uric acid production. The second is a deficiency of hypoxanthine-guanine phosphoribosyltransferase (HGPRT). HGPRT is responsible for the conversion of guanine to guanylic acid and hypoxanthine to inosinic acid. These two conversions require PRPP as the cosubstrate and are important reactions involved in the synthesis of nucleic acids. A deficiency in the HGPRT enzyme leads to increased metabolism of guanine and hypoxanthine to uric acid and to more PRPP to interact with glutamine in the first step of the purine pathway.12 Complete absence of HGPRT results in the childhood Lesch–Nyhan syndrome, characterized by choreoathetosis, spasticity, intellectual disability, and markedly excessive production of uric acid. A partial deficiency of the enzyme may be responsible for marked hyperuricemia in otherwise normal, healthy individuals.
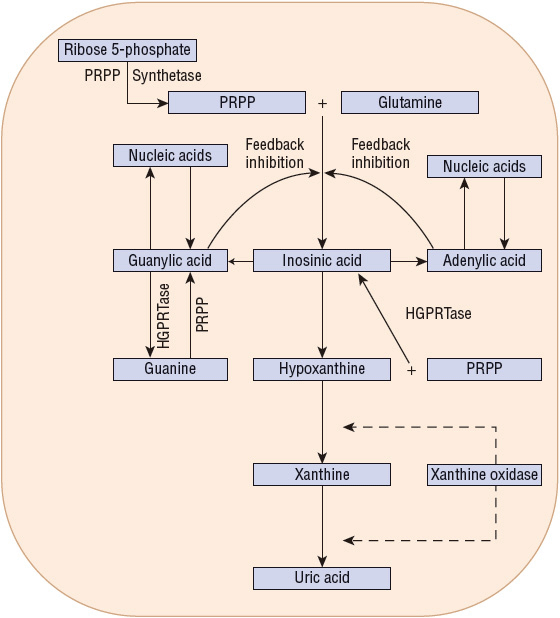
FIGURE 74-1 Purine metabolism. (HGPRT, hypoxanthine-guanine phosphoribosyltransferase; PRPP, phosphoribosyl pyrophosphate.)
CLINICAL PRESENTATION Acute Gouty Arthritis
Underexcretion of Uric Acid
Normally, uric acid does not accumulate as long as production is balanced with elimination. About two thirds of the daily uric acid production is excreted in the urine and the remainder is eliminated through the GI tract after enzymatic degradation by colonic bacteria. The vast majority of patients (90%) with gout have a relative decrease in the renal excretion of uric acid for an unknown reason (primary idiopathic hyperuricemia).2
A decline in the urinary excretion of uric acid to a level below the rate of production leads to hyperuricemia and an increased miscible pool of sodium urate. Almost all the urate in plasma is freely filtered across the glomerulus. The concentration of uric acid appearing in the urine is determined by multiple renal tubular transport processes in addition to the filtered load. Evidence favors a four-component model including glomerular filtration, tubular reabsorption, tubular secretion, and postsecretory reabsorption.
Approximately 90% of filtered uric acid is reabsorbed in the proximal tubule, probably by both active and passive transport mechanisms. There is a close linkage between proximal tubular sodium reabsorption and uric acid reabsorption, so conditions that enhance sodium reabsorption (e.g., dehydration) also lead to increased uric acid reabsorption. The exact site of tubular secretion of uric acid has not been determined; this too appears to involve an active transport process. Postsecretory reabsorption occurs somewhere distal to the secretory site. Table 74–2 lists the drugs that decrease renal clearance of uric acid through modification of filtered load or one of the tubular transport processes. By enhancing renal urate reabsorption, insulin resistance is also associated with gout.
TABLE 74-2 Drugs Capable of Inducing Hyperuricemia and Gout

The pathophysiologic approach to the evaluation of hyperuricemia requires determining whether the patient is overproducing or underexcreting uric acid. This can be accomplished by placing the patient on a purine-free diet for 3 to 5 days and then measuring the amount of uric acid excreted in the urine in 24 hours. As it is very difficult to maintain a purine-free diet for several days, this test is done infrequently in clinical practice. Nevertheless, when it is performed, individuals who excrete more than 600 mg on a purine-free diet may be considered overproducers. Hyperuricemic individuals who excrete less than 600 mg of uric acid per 24 hours on a purine-free diet may be classified as underexcreters of uric acid. On a regular diet, excretion of more than 1,000 mg per 24 hours reflects overproduction; less than this is probably normal.
CLINICAL PRESENTATION
![]() Gout is diagnosed clinically by symptoms rather than laboratory tests of uric acid. In fact, asymptomatic hyperuricemia discovered incidentally generally requires no therapy because many individuals with hyperuricemia will never experience an attack of gout. These patients should still be encouraged to implement lifestyle measures to reduce serum urate concentrations.
Gout is diagnosed clinically by symptoms rather than laboratory tests of uric acid. In fact, asymptomatic hyperuricemia discovered incidentally generally requires no therapy because many individuals with hyperuricemia will never experience an attack of gout. These patients should still be encouraged to implement lifestyle measures to reduce serum urate concentrations.
Acute Gouty Arthritis
A classic acute attack of gouty arthritis is characterized by rapid and localized onset of excruciating pain, swelling, and inflammation. The attack is typically monoarticular at first, most often affecting the first metatarsophalangeal joint (great toe) and then, in order of frequency, the insteps, ankles, heels, knees, wrists, fingers, and elbows. In one half of initial attacks, the first metatarsophalangeal joint is affected, a condition commonly referred to as podagra (see Fig. 74–2). Up to 90% of patients with gout will experience podagra at some point in the course of their disease.2
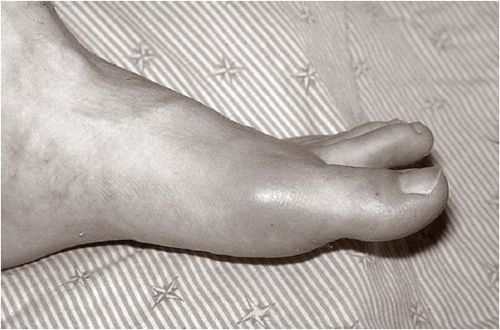
FIGURE 74-2 Acute gout attack of the first metatarsophalangeal joint. (From Imboden J, Hellmann DB, Stone JH. Current Rheumatology Diagnosis and Treatment, 2nd ed. New York: McGraw-Hill, 2004:316.)
Atypical presentations of gout also occur. For elderly patients, gout can present as a chronic polyarticular arthritis that can be confused with rheumatoid arthritis or osteoarthritis. Additionally, the onset of gout may be less dramatic than the typical acute attack and have fewer clinical findings.13 Multiple small joints in the hands may be involved, especially in elderly women.10 Table 74–3 summarizes the different clinical manifestations of gout.
TABLE 74-3 Clinical Manifestations of Gout

The predilection of acute gout for peripheral joints of the lower extremity is probably related to the low temperature of these joints combined with high intraarticular urate concentration. Synovial effusions are likely to occur transiently in weight-bearing joints during the course of a day with routine activity. At night, water is reabsorbed from the joint space, leaving behind a supersaturated solution of monosodium urate, which can precipitate attacks of acute arthritis. Attacks generally begin at night with the patient awakened from sleep by excruciating pain.
The development of crystal-induced inflammation involves a number of chemical mediators causing vasodilation, increased vascular permeability, complement activation, and chemotactic activity for polymorphonuclear leukocytes.14 Phagocytosis of urate crystals by the leukocytes results in rapid lysis of cells and a discharge of lysosomal and proteolytic enzymes into the cytoplasm. The ensuing inflammatory reaction is associated with intense joint pain, erythema, warmth, and swelling. Fever is common, as is leukocytosis. Untreated attacks may last from 3 to 14 days before spontaneous recovery.
Although acute attacks of gouty arthritis may occur without apparent provocation, a number of conditions may precipitate an attack. These include stress, trauma, alcohol ingestion, infection, surgery, rapid lowering of serum uric acid by ingestion of uric acid-lowering agents, and ingestion of certain drugs known to elevate serum uric acid concentrations (see Table 74–2). Other crystal-induced arthropathies that may resemble gout on clinical presentation are caused by calcium pyrophosphate dihydrate crystals (pseudogout) and calcium hydroxyapatite crystals, which are associated with calcific periarthritis, tendinitis, and arthritis.14–17 Acute flares of gouty arthritis may occur infrequently, but over time the interval between attacks may shorten if appropriate measures to correct hyperuricemia are not undertaken. Later in the disease, tophaceous deposits of monosodium urate crystals in the skin or subcutaneous tissues may be found. These tophi can be anywhere but are often found on the hands, wrists, elbows, or knees. It is estimated to take 10 or more years for tophi to develop.
Diagnostic Evaluation
Table 74–4 lists the differential diagnosis of an acute monoarthritis.18,19 A definitive diagnosis of gout requires aspiration of synovial fluid from the affected joint and identification of intracellular crystals of monosodium urate monohydrate in synovial fluid leukocytes.2 Identification of monosodium urate crystals is highly dependent on the experience of the observer. Crystals are needle shaped, and when examined under polarizing light microscopy, they are strongly negatively birefringent (see Fig. 74–3). Crystals can be observed in synovial fluid during asymptomatic periods.20 If an affected joint is tapped, the resulting synovial fluid may have white cells and appear purulent. Such findings should always raise the question of infection. If any clinical features of infection are present, such as high fever, elevated white blood cell count, multiple joints affected, or an identified source of infection, proper diagnosis and treatment are critical. Patients with gout can have septic arthritis. Diabetes, alcohol abuse, and advanced age increase the likelihood of septic arthritis.
TABLE 74-4 Differential Diagnosis of Acute Monoarthritis

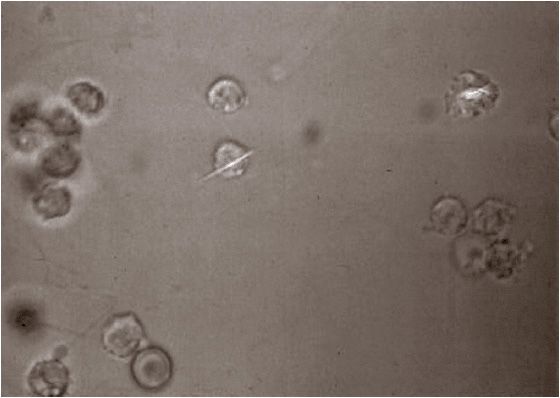
FIGURE 74-3 Urate crystal ingested by a polymorphonuclear leukocyte in synovial fluid. (From Imboden J, Hellmann DB, Stone JH. Current Rheumatology Diagnosis and Treatment, 2nd ed. New York: McGraw-Hill, 2004:317.)
In lieu of obtaining a synovial fluid sample from an affected joint to inspect for urate crystals, the clinical triad of inflammatory monoarthritis, elevated serum uric acid level, and response to colchicine can be used to diagnose gout. However, this approach has limitations, including a failure to recognize atypical gout presentations and the fact that serum uric acid levels can be normal or even low during an acute gout attack.2,5,21 In addition, use of colchicine as a diagnostic tool for gout is limited by lack of sensitivity and specificity for the disease. Other conditions such as psoriatic arthritis, sarcoidosis, and Mediterranean fever can respond to colchicine therapy. For patients with long-standing gout, radiographs may show punched-out marginal erosions and secondary osteoarthritic changes; however, in an acute first attack radiographs will be unremarkable.19,22 The presence of chondrocalcinosis on radiographs may indicate pseudogout. Some studies have recently examined the use of magnetic resonance imaging and computed tomography to obtain images for patients with gout; however, this is not currently considered part of normal practice. Table 74–5 shows the European League Against Rheumatism (EULAR) evidence-based diagnostic principles.22
TABLE 74-5 EULAR Evidence-Based Recommendations for Gout: Diagnostic Principles

Uric Acid Nephrolithiasis
Clinicians should be suspicious of hyperuricemic states for patients who present with kidney stones, as nephrolithiasis occurs in approximately 15% of patients with gout.23 The frequency of urolithiasis depends on serum uric acid concentrations, acidity of the urine, and urinary uric acid concentration. Typically, patients with uric acid nephrolithiasis have a urinary pH of less than 6. Uric acid has a negative logarithm of the acid ionization constant of 5.5. Therefore, when the urine is acidic, uric acid exists primarily in the unionized, less soluble form. At a urine pH of 5, urine is saturated at a uric acid level of 15 mg/dL (0.89 mmol/L). When the urine pH is 7, the solubility of uric acid in urine is increased to 200 mg/dL (11.9 mmol/L).1 For patients with uric acid nephrolithiasis, urinary pH typically is less than 6 and frequently less than 5.5. When acidic urine is saturated with uric acid, spontaneous precipitation of stones may occur.
Other factors that predispose individuals to uric acid nephrolithiasis include excessive urinary excretion of uric acid and highly concentrated urine. The risk of renal calculi approaches 50% in individuals whose renal excretion of uric acid exceeds 1,100 mg/day (6.5 mmol/day). In addition to pure uric acid stones, hyperuricosuric individuals are at increased risk for mixed uric acid–calcium oxalate stones and pure calcium oxalate stones. Uric acid stones are usually small, round, and radiolucent. Uric acid stones containing calcium are radiopaque.24
Gouty Nephropathy
There are two types of gouty nephropathy: acute uric acid nephropathy and chronic urate nephropathy.2 In acute uric acid nephropathy, acute renal failure occurs as a result of blockage of urine flow secondary to massive precipitation of uric acid crystals in the collecting ducts and ureters. This syndrome is a well-recognized complication for patients with myeloproliferative or lymphoproliferative disorders and is a result of massive malignant cell turnover, particularly after initiation of chemotherapy.
Chronic urate nephropathy is caused by the long-term deposition of urate crystals in the renal parenchyma. Microtophi may form, with a surrounding giant-cell inflammatory reaction. A decrease in the kidneys’ ability to concentrate urine and the presence of proteinuria may be the earliest pathophysiologic disturbances. Hypertension and nephrosclerosis are common associated findings. Although renal failure occurs in a higher percentage of gouty patients than expected, it is not clear if hyperuricemia per se has a harmful effect on the kidneys. The chronic renal impairment seen in individuals with gout may result largely from the coexistence of hypertension, diabetes mellitus, and atherosclerosis.
Tophaceous Gout
Tophi (urate deposits) are uncommon in the general population of gouty subjects and are a late complication of hyperuricemia. The most common sites of tophaceous deposits for patients with recurrent acute gouty arthritis are the base of the fingers, olecranon bursae, ulnar aspect of the forearm, Achilles tendon, knees, wrists, and hands (Fig. 74–4).2 Eventually, even the hips, shoulders, and spine may be affected. In addition to causing obvious deformities, tophi may damage surrounding soft tissue, cause joint destruction and pain, and even lead to nerve compression syndromes including carpal tunnel syndrome.
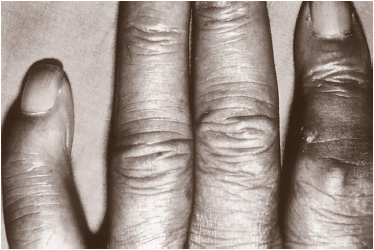
FIGURE 74-4 Tophaceous gout with subcutaneous nodule almost breaking through the skin. (From South-Paul JE, Matheny SC, Lewis EL. Current Diagnosis and Treatment in Family Medicine. New York: McGraw-Hill, 2004:275.)
TREATMENT
Desired Outcomes
The goals in the treatment of gout are to terminate the acute attack, prevent recurrent attacks of gouty arthritis, and prevent complications associated with chronic deposition of urate crystals in tissues. These can be accomplished through a combination of pharmacologic and nonpharmacologic methods, including focused patient education efforts. The first-ever American College of Rheumatology (ACR) evidence- and consensus-based guidelines for the management of gout were published in 2012.25,26 These guidelines provide specific recommendations for treatment of acute gout attacks, management of hyperuricemia in gout, and antiinflammatory prophylaxis of acute gout during initiation of urate-lowering therapy. These guidelines will be discussed throughout the remainder of the treatment section of this chapter. Tables 74–6 and 74-7 summarize dosing and monitoring information for available pharmacotherapy used in management and prevention of gout.
TABLE 74-6 Pharmacotherapy of Acute Gout, Antiinflammatory Prophylaxis During Initiation of Urate-Lowering Therapy and Hyperuricemia in Gouta
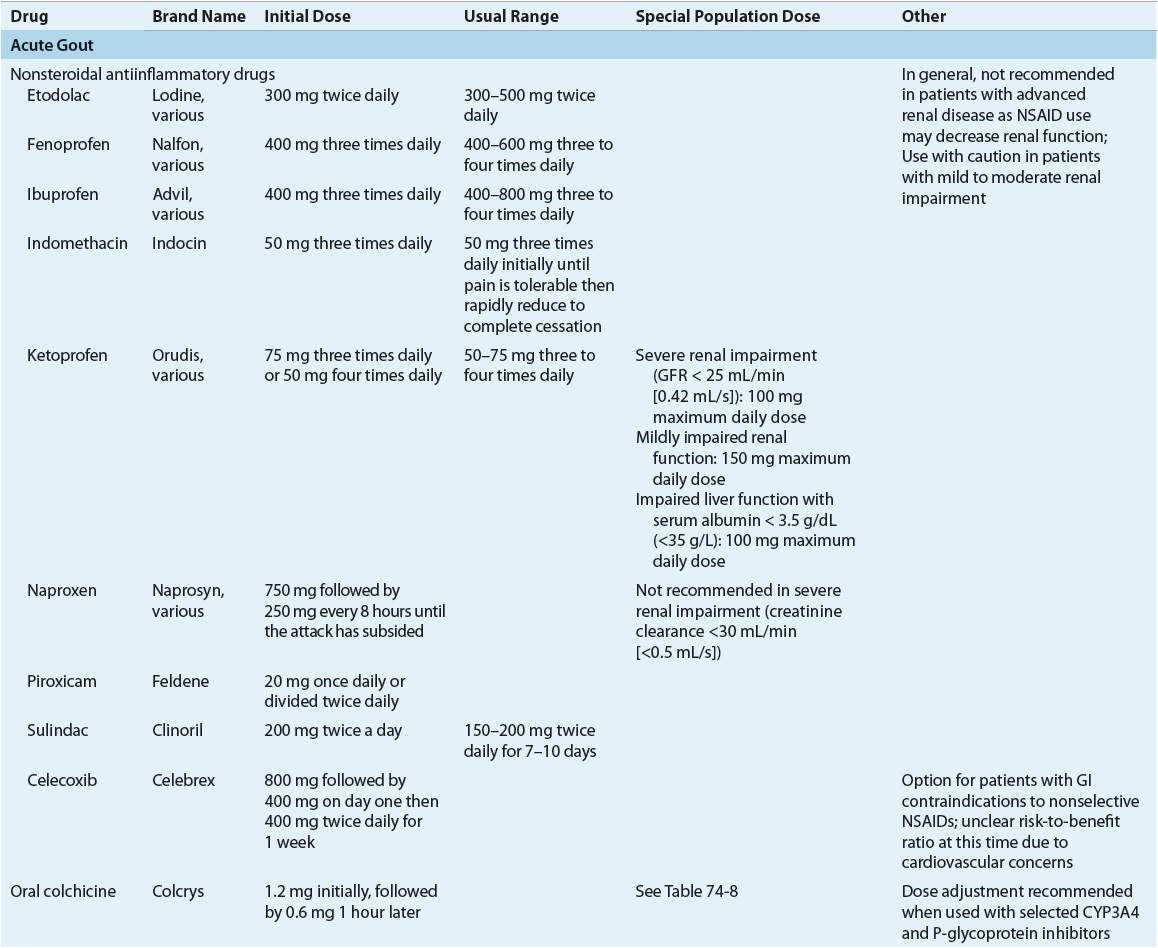

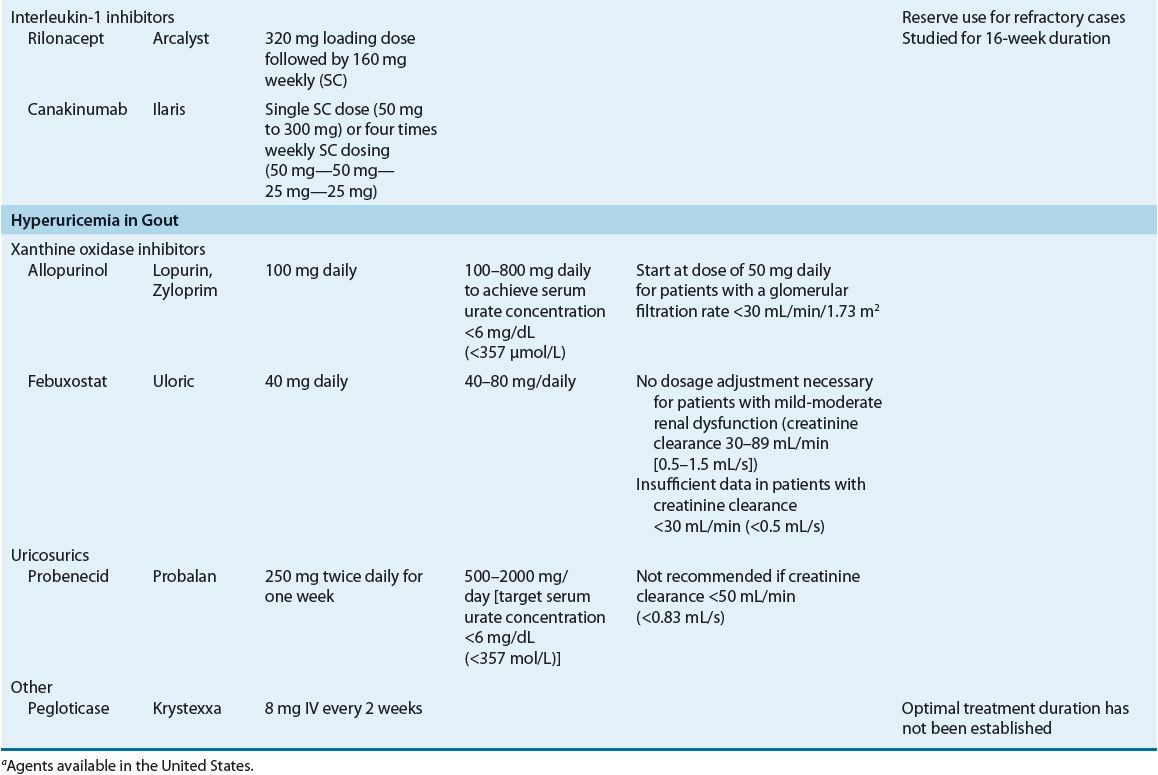
TABLE 74-7 Drug Monitoring


Acute Gouty Arthritis
Nonpharmacologic Therapy
There are limited effective nonpharmacologic therapies for an acute gout attack; therefore, they are recommended strictly as adjunctive treatment.
Local ice application is the most effective.26 In one small study, adjunctive ice application resulted in significantly greater pain reduction in those receiving the therapy compared with those not treated with ice (difference of 3.33 cm on a 10-cm visual analog pain scale, P = 0.021).27 Complementary and alternative medicines, including flaxseed and celery root, are not recommended in ACR guidelines.26
Pharmacologic Therapy
![]() For most patients, acute attacks of gouty arthritis may be treated successfully with nonsteroidal antiinflammatory drugs (NSAIDs), corticosteroids, or colchicine. The ACR guidelines recognize these three modalities as first-line monotherapy for the treatment of acute gout. Treatment should commence within 24 hours of the onset of an attack. In more severe cases, those affecting multiple joints or causing higher intensity pain, combination or investigational drug therapy may be indicated (Fig. 74–5).26
For most patients, acute attacks of gouty arthritis may be treated successfully with nonsteroidal antiinflammatory drugs (NSAIDs), corticosteroids, or colchicine. The ACR guidelines recognize these three modalities as first-line monotherapy for the treatment of acute gout. Treatment should commence within 24 hours of the onset of an attack. In more severe cases, those affecting multiple joints or causing higher intensity pain, combination or investigational drug therapy may be indicated (Fig. 74–5).26
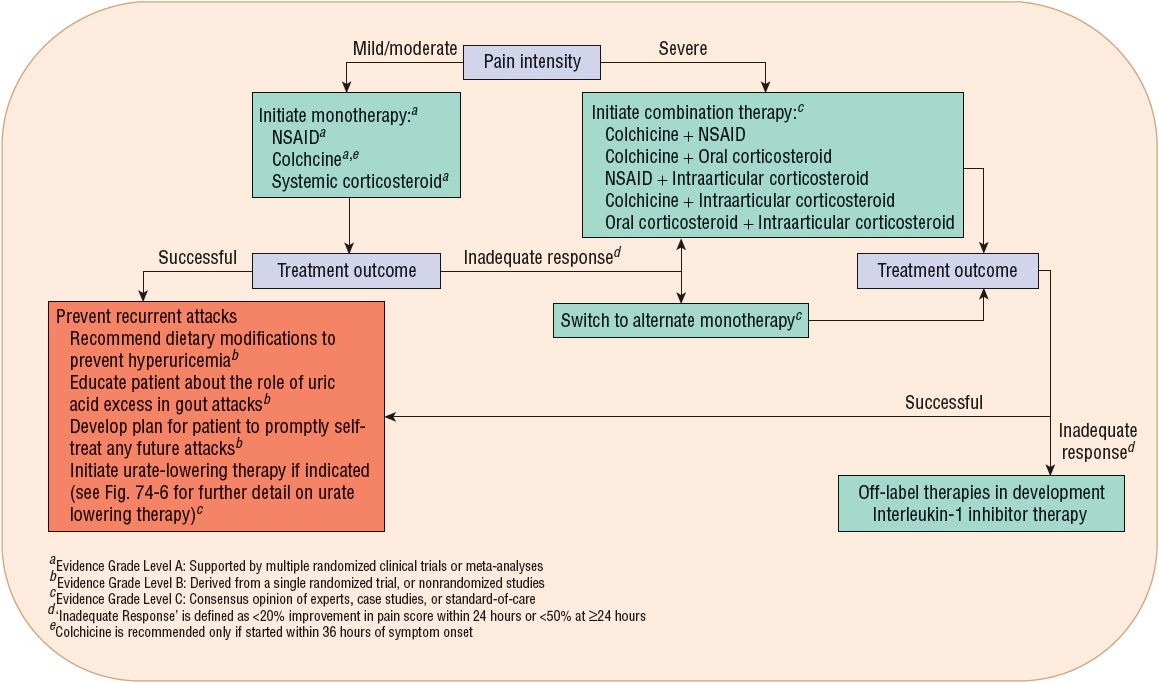
FIGURE 74-5 Algorithm for management of an acute gout attack.
Nonsteroidal Antiinflammatory Drug
NSAIDs are a mainstay of therapy for acute attacks of gouty arthritis because of their excellent efficacy and minimal toxicity with short-term use. Indomethacin has been historically favored as the NSAID of choice for acute gout flares, but there is little evidence to support one NSAID as being more efficacious than another. Three agents (indomethacin, naproxen, and sulindac) have U.S. Food and Drug Administration (FDA)-approved labeling for the treatment of gout, although several others are likely to be effective.26 Although choice of NSAID is not an important determinant of therapeutic success, timing of pharmacotherapy is. It is critical that therapy is initiated within 24 hours of acute gout attack onset and continued until complete resolution.26 Following resolution of the attack, tapering of NSAID therapy may be considered, especially in patients with comorbidities such as hepatic or renal insufficiency where prolonged therapy would be undesirable.26 Resolution of an acute attack for most patients generally occurs within 5 to 8 days after initiating therapy.
All NSAIDs have the potential to cause similar adverse effects. The most common areas affected include the GI system (gastritis, bleeding, perforation), kidneys (renal papillary necrosis, reduced creatinine clearance), cardiovascular system (sodium and fluid retention, increased blood pressure), and CNS (impaired cognitive function, headache, dizziness). Caution should be exercised when using NSAIDs for individuals with a history of peptic ulcer disease, congestive heart failure, uncontrolled hypertension, renal insufficiency, coronary artery disease, or who are concurrently receiving anticoagulants or antiplatelets. Patients with active peptic ulcer disease, uncompensated congestive heart failure, severe renal impairment, or a history of hypersensitivity to aspirin or other NSAIDs should not be prescribed an NSAID.
Selective cyclooxygenase-2 (COX-2) inhibitors present a potentially better tolerated alternative to nonselective NSAIDs in patients with GI issues. Specific COX-2 inhibitors, etoricoxib and lumiracoxib, have demonstrated efficacy in the treatment of acute gout in numerous controlled trials; however, these agents are not available in the United States. One study has established effectiveness of high-dose celecoxib (1200 mg on day 1 followed by 400 mg twice daily thereafter) in the treatment of acute gout, but concerns regarding the cardiovascular risk of COX-2 inhibitors must be considered when using these agents (see Chap. 71, Osteoarthritis, for further discussion of COX-2 inhibitors).28,29 The ACR guidelines recommend celecoxib as an option for patients unable to take NSAIDs but note that the risk-to-benefit ratio of celecoxib use in acute gout is unclear.26
Corticosteroids
Corticosteroids have historically been reserved for treatment of acute gout flares when contraindications to other therapies exist, largely due to lack of evidence from controlled clinical trials. However, more recent evidence indicates that corticosteroids are equivalent to NSAIDs in the treatment of acute gout flares.30 They can be used either systemically or by intraarticular injection. The ACR guidelines recommend that the number of joints involved be considered when choosing the route of corticosteroid administration. If only one or two joints are involved, either intraarticular or oral corticosteroids are recommended. If an attack is polyarticular, systemic therapy is necessary.26 A hypothetical risk for a rebound attack upon steroid withdrawal exists; therefore, gradual tapering is often employed when discontinuing steroid therapy. The ACR guidelines suggest two different dosing strategies for oral corticosteroid therapy (prednisone or prednisolone) in the treatment of acute gout: (a) 0.5 mg/kg daily for 5 to 10 days followed by abrupt discontinuation or (b) 0.5 mg/kg daily for 2 to 5 days followed by tapering for 7 to 10 days. The guidelines also support the use of a methylprednisolone dose pack for acute treatment of gout, a 6-day regimen that starts with 24 mg on day 1 and decreases by 4 mg each day.26 Intraarticular administration of triamcinolone acetonide in a dose of 20 to 40 mg may be useful in treating acute gout limited to one or two joints. Injection should be done under an aseptic technique in a joint determined not to be infected. Per ACR guideline recommendations, intraarticular corticosteroid therapy should be used in conjunction with either an NSAID, colchicine, or oral corticosteroid therapy; however, case reports suggest that this therapeutic approach may be as effective as monotherapy.26,31 A single intramuscular injection of a long-acting corticosteroid, such as methylprednisolone, followed by oral corticosteroid therapy is recognized as a reasonable therapeutic approach to the treatment of acute gout by the ACR guidelines.26 Alternatively, intramuscular corticosteroid monotherapy may be considered in patients with multiple affected joints who are unable to take oral therapy.
The adverse effects of corticosteroids are generally dose and duration dependent. Short-term use for treatment of acute attacks is generally well tolerated. Corticosteroids should be used with caution for patients with diabetes as they can increase blood sugar. In addition, patients with a history of GI problems, bleeding disorders, cardiovascular disease, and psychiatric disorders should be monitored closely. Long-term corticosteroid use should be avoided because of the risk for osteoporosis, hypothalamic–pituitary axis suppression, cataracts, and muscle deconditioning that can occur with their use.
Corticotropin, or adrenocorticotropic hormone (ACTH), which stimulates the adrenal cortex to produce cortisol and corticosterone, can be administered in acute gout. Doses of 40 to 80 United States Pharmacopeia (USP) units are given intramuscularly every 6 to 8 hours for 2 to 3 days, and then discontinued. Studies with ACTH are limited, but it appears to provide similar efficacy to systemic antiinflammatory doses of corticosteroids.32 When administered alone or in combination with colchicine, ACTH may provide earlier efficacy compared with indomethacin but with fewer adverse effects.33 Because the studies have several limitations, the regimen should be considered only as an alternative, especially for patients with comorbidities where other regimens are contraindicated.34 Examples of patients where ACTH has been used safely when other first-line gout therapies were contraindicated include those with congestive heart failure, chronic renal failure, and history of GI bleeding.35 The ACR guidelines support the use of ACTH in the treatment of acute gout in patients unable to take oral medications.26
Colchicine
![]() Colchicine is an antimitotic drug that is highly effective at relieving acute attacks of gout.36 When begun within the first 24 hours of an acute attack, colchicine produces a response in two thirds of patients within hours of administration.37 If the initiation of colchicine is delayed; however, the probability of success with the drug diminishes substantially. For this reason, the ACR guidelines advocate use of colchicine for treatment of acute gout only if started within 36 hours of attack onset.26
Colchicine is an antimitotic drug that is highly effective at relieving acute attacks of gout.36 When begun within the first 24 hours of an acute attack, colchicine produces a response in two thirds of patients within hours of administration.37 If the initiation of colchicine is delayed; however, the probability of success with the drug diminishes substantially. For this reason, the ACR guidelines advocate use of colchicine for treatment of acute gout only if started within 36 hours of attack onset.26
Although it is a highly effective therapy, oral colchicine can cause dose-dependent GI adverse effects, including nausea, vomiting, and diarrhea. Other important non-GI adverse effects include neutropenia and axonal neuromyopathy, which may be worsened for patients taking other myopathic drugs such as β-hydroxy-β-methylglutaryl-coenzyme A reductase inhibitors (statins) or for those with renal insufficiency.
Colchicine was used for many years as an unapproved drug with no FDA-approved prescribing information, dosage recommendations, or drug interaction warnings. More recently, the FDA approved a 0.6-mg tablet of colchicine (Colcrys®) for oral use. Data submitted in support of the safety and efficacy of colchicine in acute gout flares demonstrated that a substantially lower dose of colchicine (1.2 mg initially, followed by 0.6 mg 1 hour later) was as effective as higher doses traditionally used (continued hourly dosing until symptoms subside or GI symptoms become intolerable).38 These findings suggest that prior use of high-dose colchicine regimens, may unnecessarily expose patients to increased toxicity with no additional efficacy. In addition to the new low-dose regimen, the ACR guidelines also suggest that colchicine 0.6 mg once or twice daily can be started 12 hours following the initial 1.2 mg dose and continued until the acute attack resolves.26 This off-label dosing recommendation is based upon pharmacokinetic data that suggests that colchicine levels begin to decline 12 hours after administration.38
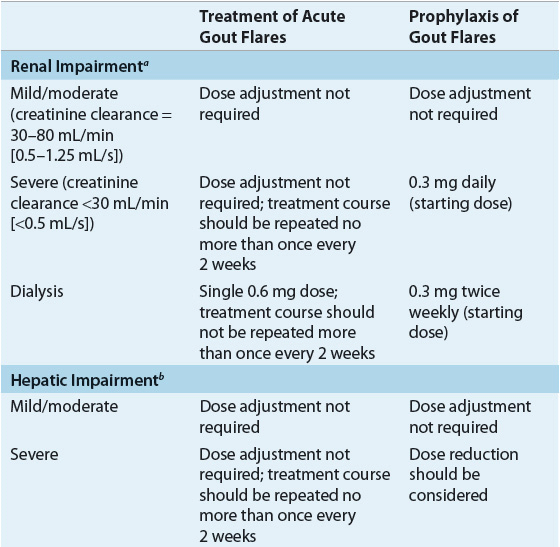
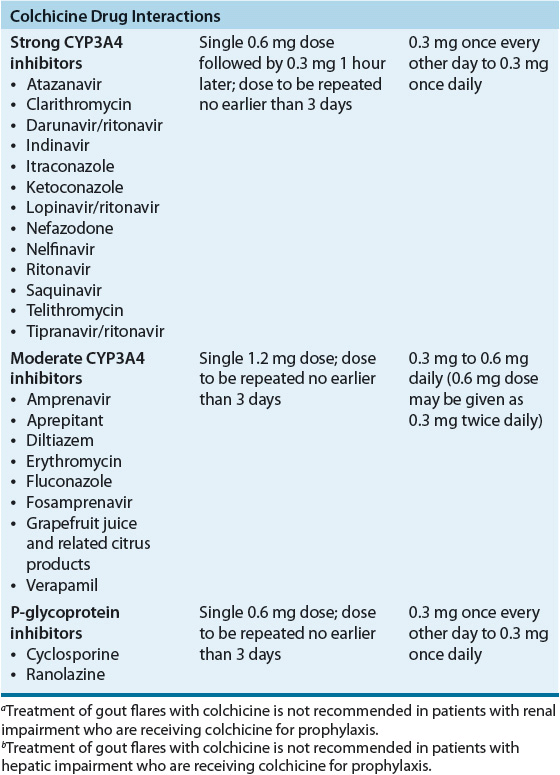
Comprehensive review of postmarketing safety data revealed an increased risk of adverse events for patients receiving colchicine administered concurrently with P-glycoprotein or cytochrome P450 3A4 inhibitors (e.g., clarithromycin or cyclosporine) (Table 74–8).39–42 These interactions are thought to result in an increased colchicine concentration. Colchicine should also be used carefully for patients with renal and hepatic insufficiency. Refer to Table 74–8 for colchicine dosing recommendations in these special situations.
TABLE 74-8 Colchicine Dosing in Special Situations/Colchicine Drug Interactions