- Although DNA replication is usually carried out with fastidious precision, copying errors will creep in all the same simply because of the enormity of the task.
- Substantial DNA damage is also engendered under the combined auspices of extrinsic factors, such as ultraviolet (UV) light, ionizing radiation, and other genotoxic agents, that connive with radical elements from within, such as reactive oxygen species (ROS), to damage our DNA.
- Without efficient processes to monitor for and then repair the damaged or inaccurately copied DNA, our cells would not survive. From this, we can conclude that DNA damage responses (DDRs) are able to dramatically reduce the accumulation of damaged DNA even if they cannot prevent it entirely.
- Evolution and cancer may follow because, under some circumstances, DNA replication is sanctioned even though the DNA is improperly repaired. As a consequence, mutations are passed on to successive generations of:
- An unresolved conundrum is why these cellular pariahs, with their potentially threatening unrepaired DNA damage, do not opt for one of the quixotic failsafe responses at their disposal and either commit suicide or permanently renounce replication. Moreover, the means by which a cell negotiates this final dilemma, apoptosis or senescence, are not fully understood.
- Another question that has long vexed cancer biologists is whether mutations arise by chance (stochastically) and are then selected for, or whether cancer development is consequent upon first acquiring a propensity to develop mutations. The answer for different cancers is likely a combination of both at different stages of progression.
- Aberrant DDRs arising through epigenetic or genetic changes may be present in premalignant colon cancer lesions, suggesting that, in this example of a multistage cancer pathway, a propensity to “epi-mutations” may come first.
- Sundry “caretaker genes” have evolved to tackle the surveillance and maintenance of DNA integrity; loss of these genes will give rise to cell death or to cells that survive with genomic instability and defective mismatch repair – a major cause or effect of carcinogenesis. In fact, around 200 proteins are now known to be involved in DDRs, and have to deal with substantive DNA damage and also an average point mutation rate of around 1.5 × 10−10 per base pair per cell generation.
- The caretaker genes are major barriers to the initiation and progression of cancer and, alongside the oncogenes and tumor suppressor genes, represent the third major class of genes subject to mutations and epigenetic silencing in cancer.
- Several, and arguably the most clinically important, hereditary cancer syndromes result from inactivating genes involved in DDR; these include, to name a few, BRCA1 and BRCA2 or PALB2 in hereditary breast cancers, ATM in ataxia telangiectasia, and various mismatch repair (MMR) genes in Lynch syndrome–related colorectal cancers.
- DNA damage is either cytotoxic or mutagenic. Thus, severely damaged DNA, as exemplified by interstrand crosslinks and double-strand breaks (DSBs) that are not successfully repaired, will culminate in cell death because normal transcription, replication, and chromosome segregation are prevented, whereas lesser endurable damage that is not corrected before replication could cause miscoding resulting in mutations and subsequent carcinogenesis.
- By implication, the more extreme the degree of DNA damage that a cell can survive, the greater the risk of propagating a cancer-causing mutation.
- Conversely, apoptosis and senescence represent a deus ex machina if the situation cannot be resolved in any other way.
- DNA damage-signaling mechanisms seem to have evolved primarily to respond to cytotoxic lesions (and many of these overlap with those involved in telomere maintenance; see Chapter 9), whereas DNA repair mechanisms have evolved to deal with both classes of DNA lesions.
- Loss of telomeres results in tumors acquiring chromosomal re-arrangements with amplification and deletion of chromosomal regions (Chapter 9).
- DNA damage sensing likely takes place primarily during replication or transcription as the DNA strands may be more readily accessible to key “sensing” proteins at these times.
- DNA damage-signaling proteins, which are activated following detection of DNA damage, are the panjandrums of proliferation and are responsible for keeping damaged cells arrested at specific cell-cycle stages so that cells can attempt DNA repair before they are allowed to proceed to the S phase or mitosis.
- Once the DNA has been repaired and the crisis has blown over, replication can resume unabated. However, if effective DNA repair is impossible or damage is extensive, these signaling processes can also trigger irreversible growth arrest or apoptosis of the cell (see Chapters 8 and 9). The p53–p21Cip1 tumor suppressor pathway plays a central role in this.
- Recent work suggests that the DDR may also be a factor in regulating cell differentiation in stem cells.
- Four main types of DNA damage are recognized: base modification, often by methylation and de-amination; mismatches due to defective proofreading during replication of DNA; breaks in the DNA backbone (single-strand breaks (SSBs) or DSBs); and crosslinking between bases on the same DNA strand or adjacent strands.
- Assorted DNA damage repair mechanisms have been identified, but there is considerable overlap between them, and which is favored in a particular damage scenario is not always predictable: the simplest employs glycosylases and other enzymes to correct aberrant methylation and de-amidation; the remaining require excision of a damaged region and then repair, and these include base excision repair (BER), nucleotide excision repair (NER), and mismatch repair (MMR). DSBs are difficult to repair and require additional mechanisms because there is no intact template strand from which to restore a correct DNA sequence.
- DSBs are thus repaired by two major processes:
- DNA mismatch repair (MMR) helps maintain genome stability by correcting DNA replication errors. MMR is initiated by mutSalpha (mutSα) or MutSbeta (mutSβ) dimers binding to mismatched DNA. Loss of MMR can drive a mutator phenotype and genetic instability. MMR increases the fidelity of DNA replication about 1000-fold.
- Although discussed in more detail in Chapter 11, ubiquitination (ubiquitylation) appears to be a key regulatory step in the recruitment of a variety of DNA repair proteins, including the core histone H2A and its variant H2AX in DSB repair.
- In response to diverse stresses, exemplified by DNA damage, the well-known tumor suppressor p53 prevents propagation of a potential cancer cell by inducing genes which arrest the cell cycle or promote apoptosis. Interestingly, p53 may also regulate genes required for successful DNA repair and moreover can reduce levels of ROS, thus protecting the genome from damage. This duality of p53 roles, as both protector and executioner, may in part be explained by the differences in genes regulated by p53 at low levels and at persistent or high levels, respectively. At low levels and early after activation, p53 mediates growth arrest, survival, and DNA repair, whereas high or persistent p53 activation mediates increased levels of p21Cip1, ROS, and apoptosis.
- Failure of DNA damage responses, due to epi-mutations in “caretaker genes” or tumor suppressors, will allow the inappropriate survival and replication of cells with damaged DNA.
- Thus, once the usually impeccable fidelity of DNA replication is vitiated by, among other things, defective mismatch repair, further mutations can accumulate at a preternatural rate. This is a cardinal feature of cancer cells, culminating in one of the two major subtypes of genomic instability:
Introduction
Human life … is a sort of target – misfortune is always firing at it, and always hitting the mark.
Wilkie Collins
Given the intimate relationship between cell-cycle and DNA damage surveillance and repair (and, when this fails, DNA mutations), there is considerable overlap between this chapter and Chapters 3 and 4. Moreover, DNA damage represents the driving force behind tumorigenesis, and as such must contend with various cancer-restraining barriers such as apoptosis and senescence, which comprise the focus of Chapters 7, 8, and 9. With this comes a degree of repetition but with a different emphasis – we make no apology for this but ask you to indulge us as these are, after all, the central processes in tumorigenesis, and they are inextricably linked. This chapter will focus more on DNA damage and its repair and less on how the cell cycle is stalled in order for this to take place or what happens to cells if repair fails – this has been the focus of other chapters.
With around 2 m of DNA each, the 1014 cells that constitute the adult human contain a total length of DNA that if stitched end to end would be around 200 000 000 000 (that’s 200 billion) km long and comprise a stretch of 1024 bases (6 billion base pairs per cell in the human genome), all subject to daily threat of damage and mutation. The Herculean task of patrolling this DNA, identifying damage, and then repairing it is delegated to each individual cell, which has to answer for its own 2 m and around 6 × 109 bases. Given that to all intents and purposes, all diploid cells contain the same DNA as each other, it might seem wasteful that this task could not in some way be coordinated between multiple cells. Well, in one circuitous way it is. DNA damage is most dangerous during cell replication (also, because DNA is unraveled, this is usually when it is detected) as the damage could be propagated, whereas even fatal damage to the DNA of most cells that cannot replicate is largely irrelevant to the organism – you have a myriad more cells, and the odd bad apple can usually be replaced. However, propagation of DNA damage can give rise to a new clone of cells that, if expanded, could compromise the survival of the whole organism. Coordination arises indirectly by means of one of the most startling examples of selfless cellular behavior observed in the context of a multicellular organism. Namely, a cell that determines that it has too much DNA damage to repair effectively either commits suicide or permanently declines to reproduce itself in order to serve the greater good – a cell will sacrifice itself rather than threaten the existence of the whole organism. In either case, the privilege of producing a new cell of the required type now falls to another cell without DNA damage, as long as it too can avoid and/or circumvent the various checkpoints already discussed in Chapter 4, a perfect outcome for the organism – usually. Not surprisingly, defects in any of these processes (detection, repair, apoptosis, or growth arrest) can result in cells with DNA damage replicating and propagating the defects – the essential platform for cancer.
It is always worth restating a key fact, and it is without apology that we repeat the mantra that DNA damage is unavoidable. Under the influence of extrinsic and intrinsic insults, mammalian cells are continuously acquiring DNA lesions. Extrinsic factors include exposure to UV light, anticancer drugs, and ionizing radiation, and intrinsic factors include reactive oxygen species (ROS), replication errors, and stalled replication forks. You can avoid some of the extrinsic causes, or mitigate them with sun cream, but nothing can prevent simple chance errors during DNA replication. On average we experience DNA damage at a rate between about 10 000 and 500 000 bases per cell per day, mainly due to chemical changes such as base loss by deamination or depurination and through oxidative damage. Fortunately, fewer than one in a thousand become permanent, because DNA repair mechanisms ensure that the genome remains intact, and normal cells can prevent the occurrence of mutations at the nucleotide sequence level and the chromosome level. These mechanisms include enzymes that repair damaged DNA, and signal transduction pathways (checkpoints) that induce cell-cycle arrest or apoptosis when individual stages in the cell cycle are not appropriately completed. In addition, cells with chromosomes not properly attached to the mitotic spindles are prevented from undergoing mitosis. In contrast to normal cells, most tumor cells acquire genomic instability resulting in multiple mutations – some of which may promote tumor development. The gene products that control genomic stability in normal cells have been defined to a certain extent, some of which will be discussed in this chapter. Not surprisingly, perhaps, defects in those genes that control genomic stability can contribute to tumor development.
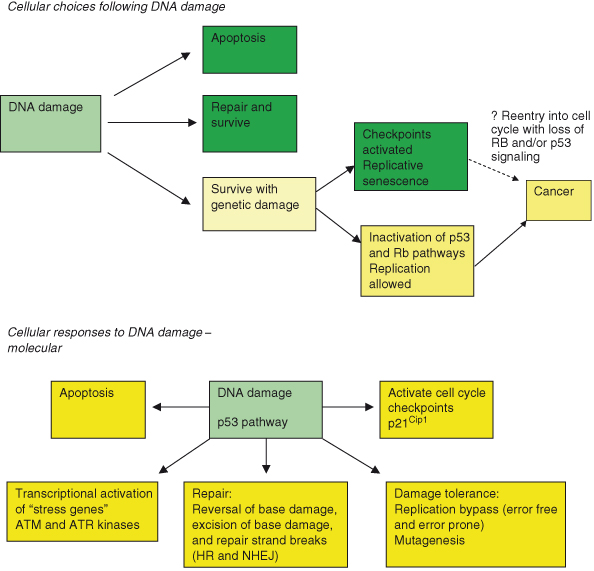
DNA damage triggers a byzantine complex of signaling cascades which may be divided into (a) DNA damage sensors, (b) checkpoint transducers, (c) checkpoint mediators, and (d) checkpoint effectors. The checkpoints that are activated during the cell cycle when mutations are detected were discussed in depth in Chapter 4. Through the operation of these checkpoints, cells with damaged DNA are blocked from cell-cycle progression and DNA replication, DNA damage repair (DDR) processes can be recruited, and the damage is repaired. When DNA damage affects only one strand, then repair is far simpler as the normal strand can be used as a template, a nucleotide crib sheet, to make certain that the repair is accurate. If both strands are damaged, as in double-strand breaks (DSBs), then a more convoluted series of repair steps is required. In fact, the pervasiveness and variety of DNA damage on offer have resulted in the evolution of highly sophisticated systems to replicate and repair DNA accurately and efficiently or, if this fails, a variety of “damage limitation” mechanisms will kick in (Fig. 10.1). In other words, if the DNA cannot be fixed, then at least the potential cancer risk can be removed by the suicide or “celibacy” of the damaged cell. This does, however, leave the cell on the horns of a dilemma, the resolution of which may have profound implications to cancer biology and the development of targeted therapies. Dead cells cannot pose any future risk of cancer but must be replaced at some energy cost to the organism, whereas growth-arrested, or celibate, ones could conceivably recover their lost appetites over time.
Figure 10.1 Cellular responses to DNA damage. Following DNA damage, the cell may undergo one of three fates: repair and survive, fail to repair, and apoptosis or replicative senescence. Rarely the cell with damaged DNA may still be able to replicate and thus pose a threat of cancer. The molecular processes activated by DNA damage are shown in the second part of the figure.
A complete set of chromosomes in a human somatic cell contains around 6 × 109 base pairs of DNA. Although some of these comprise sequences representing genes and their regulatory elements, large stretches of DNA appear not to encode anything and are particularly prone to mutation (though, as discussed, some of this erstwhile “Junk” DNA is now known to encode, among others, microRNAs (miRNAs) that are subject to cancer-relevant mutations). The forces aligned against our genomes are legion, and DNA in every cell of our bodies is spontaneously damaged thousands of times every day. Specific sequences are particularly prone to this. For example, some repetitive DNA tracts such as microsatellites and mini-satellites are naturally unstable and, in humans, alterations in these sequences are often associated with genetic diseases and cancer. There are many different types of DNA damage, including: conversion of bases by loss of an amino group (thus cytosine can be converted to uridine), mismatches during DNA replication (such as incorporation of uridine instead of thymine), breaks in the DNA backbone (either SSBs or DSBs), and crosslinking between bases on the same or opposite strands (may also be induced by chemotherapy).
To some extent, extrinsic DNA-damaging agents may be avoided or exposure to them minimized (sun screens and sun avoidance spring to mind). Importantly, it may also be possible to minimize the damage caused by some intrinsic factors; thus, in part the development of ROS may be reduced by healthy lifestyle choices, including diets low in saturated fat and cholesterol and high in anti-oxidants – dietary modifications are also convincingly linked to reducing risk of coronary heart disease. However, given that oxidative metabolism is a “nonnegotiable” and pivotal aspect of the life of a eukaryotic cell, any processes favoring release of superoxides from mitochondria may culminate in DNA damage, no matter how healthy the diet. During tumorigenesis, processes both intrinsic and extrinsic to the cancer cell can provoke oxidative stress. Thus, deregulated expression of oncogenes such as c-MYC can promote mitochondrial biogenesis and formation of ROS, which may be a contributory factor in the well-recognized propensity of oncogenic MYC to promote genomic instability. Recent studies suggest that neighboring stromal cells might indirectly provoke DNA damage in adjacent nascent cancer cells; matrix metalloproteinsases (MMPs) released by stromal cells may induce release of ROS from mitochondria in cancer cells.
Given that the DNA in a diploid human cell is very long (about 2 m in length), very convoluted, and very compacted (packaged into a compact chromatin structure by histones), monitoring and repairing damaged DNA are not trivial undertakings and involve a large and ever-expanding group of proteins encoded by what have been dubbed “caretaker” genes. But in an archetypal “snake eats tail” conundrum, these caretaker genes are themselves subject to mutations and epigenetic silencing. The resultant disruption of mechanisms that regulate cell-cycle checkpoints, DNA repair, and apoptosis result in genomic instability – a major contributor to tumorigenesis. Moreover, DNA damage responses take place within the context of the cell cycle and therefore affect and integrate with various checkpoints discussed in Chapters 4, 7, and 8. Not surprisingly, therefore, many tumor suppressor pathways, frequently subject to “epimutations” in cancer, such as the ARF–p53–p21Cip1 and p16INK4a–RB pathways, are also involved in DDR, alongside the caretaker gene products more specifically dedicated to this task.
Despite considerable redundancy and cross-talk, there are broadly two major pathways, plus a more recent addition, activated by different types of DNA damage:
- The ATR–CHK1 pathway is triggered by a wide variety of insults, including but not restricted to single-stranded DNA and stalling of DNA replication forks. Specific detection of relevant DNA damage falls to the ATRIP protein and the RAD9–RAD1–HUS1 cell-cycle checkpoint complex (9-1-1 complex), which forms a clamp-like complex that interacts with and activates proteins involved in base excision repair (BER) as well as with various checkpoint proteins. ATM–CHK1 is also recruited during most other forms of DDR at some point.
- The ATM–CHK2 (and parallel DNA-PKcs) pathway is triggered primarily by DNA DSBs, which are sensed by as-yet-unconfirmed means but likely include the MRE11–RAD50–NBS1 complex (MRN complex) and KU80 protein.
- A MAPKAP kinase 2 (MK2) regulated pathway has been identified recently.
There are numerous examples of cross-talk, and it appears increasingly likely that the ATR–CHK1 pathway is in some way involved in most (if not all) DDRs. Thus MRN may activate some ATR responses, and it appears that, following initial ATM–CHK2 activation in DSB repair, ATR is also activated. It is worth noting that recognition of DSBs poses certain problems for the DDR; DSBs occur naturally during meiosis, so how do the ends of chromosomes avoid being recognized as a DSB, particularly in light of the presence of proteins involved in DDR, such as KU, at the telomere (discussed in Chapter 9).
The sensors (MRN and 9-1-1) detect DNA damage and recruit the transducers, ATM and ATR, which in turn activate further transducers, CHK2 and CHK1, respectively. ATM–ATR activation allows recruitment and activation of a plethora of mediators, including H2AX, 53BP1, Claspin, BRCA1, TopBP1, MDC1, SMC1, FANCD2, Timeless, and Tipin. These remain rooted to the site of damage, whereas CHK1 and CHK2 are liberated to unleash downstream signaling. The net result is the shackling of the cell cycle at several distinct phases by inactivation of CDC25s and/or activation of p53 effectors. Some of the complexity, the sequential rubs and bumps of partner proteins as they dance around the broken DNA, has been captured. Thus, RPA attaches to the damaged area and is joined by Timeless and Tipin, thereby enabling Claspin to associate with and phosphorylate CHK1, which in turn allows BRCA1 to join the melée. TopBP1 directly activates ATR–ATRIP and promotes ATR-mediated CHK1 phosphorylation.
ATM–ATR pathways appear to operate on two different time scales. Thus, growth-stalling inhibition of CDC25s proceeds rapidly, whereas the potentially more lethal p53-dependent pathway, which may be preferentially activated by nastier and difficult-to-fix DNA damage, is slower. This provides a period of respite during which repairs may be carried out before the suicidal urge becomes irresistible. Numerous other effectors have been identified, including a variety of kinases and repair proteins.
CHK1 can activate DNA repair kinases such as DNA-PKcs, which partners Ku70 and Ku80 in DSB repair. Phosphorylation of FANCE and Rad51 by CHK1 is needed for Fanconi anemia (FA)/BRCA-directed and homologous repair, respectively.
Genomic instability and cancer are held at bay by a series of different repair systems, but two are most frequently associated with cancer:
- The DNA mismatch repair (MMR) system, which corrects DNA sequence errors generated during DNA replication, during recombination, and by mutagenic agents, such as the drug cis-platinum;
- The repair of DNA DSBs, induced by ionizing radiation and drugs such as etoposide.
In fact, like ebbing tides and receding waves, small point mutations are eliminated by postsynthesis MMR, thereby improving the fidelity of DNA replication by several orders of magnitude (more than 100-fold!). DSBs are amongst the most indomitable of all DNA damage, and much space will be devoted to these in the chapter. Various proteins involved in mismatch and DSB repair also act to couple DNA repair to cell-cycle checkpoint regulation and apoptosis. It is worth noting here that promoting extreme genetic damage and instability with drugs or radiation may also paradoxically be a means of treating cancer; this is effective as long as growth arrest and/or apoptosis of cancer cells are appropriately triggered as a result. Not surprisingly, given everything that has been discussed regarding cancer cells so far, cancer cells are singularly accomplished at upregulating the efficiency of their DNA repair systems, thereby making them more capable of resisting DNA-damaging therapies. Both the ATM–CHK2 and ATR–CHK1 checkpoint pathways are activated after treatment with DNA-damaging agents; the relative contributions depend both on the agents used and on the nature of the damage ensuing. The result for the cell is either cell-cycle arrest (in the G1, S, or G2 and M phases) or death. These outcomes are all driven by various targets of the ATM–ATR kinases and include the tumor suppressor p53 (and in turn key p53 targets, in particular p21CIP1 for inducing growth arrest in G1, and BAX, PUMA, and NOXA for promoting apoptosis) and the proapoptotic BCL-2 family member BID, which may mediate S-phase arrest (alongside various other ATM–ATR targets) as well as contribute to cell death and inhibition of CDC25, which causes arrest at the G2 or M phase. Coordinated action between proteins involved in cell-cycle arrest, apoptosis, and DNA repair is evident at many levels, suggesting that a delicate balance between factors dictates if a cell with damaged DNA survives and repairs this or undergoes apoptosis, presumably with the survival option reviewed at various points in the process.
In the unlikely eventuality that you have somehow skipped over this, cancers are generally accepted as clonal in origin with the cancer cell arising through the stepwise accumulation of gene alterations in a process akin to Darwinian evolution and natural selection. At least for some cancers, it has been suggested that the iterative process of mutation and clonal evolution driving tumorigenesis may be compounded by various factors that together greatly increase the propensity of such nascent cancer cells to mutate. In fact, the prior acquisition of genomic instability is regarded by some as a condicio sine qua non in the progression of human cancer, because only then can mutations in other genes take place at a sufficient rate. It is worth noting that although cancers may be clonal, their chromosomes and genes may not be – in other words, genes and chromosomes may be quite heterogeneous, largely due to their inherent genomic instability.
However, the role of genomic instability in initiating cancers is still not fully elucidated. What is generally uncontested is that the majority of human cancers, when studied, exhibit genomic instability. But when in the life of the cancer has this occurred, and is it a cause or effect of cancer? Possibly the best studied with regard to the role of genomic instability in patho-etiology are colorectal and breast cancers (discussed in this chapter). A number of recent studies using in vitro and in vivo models of breast cancer, among others, suggest that genomic instability (in some cases related to telomere shortening – see Chapter 9) might take place early or at the transition from benign to malignant cancer. Last year, two research projects employed sequencing to look at clonal populations in different cancers and concluded that genomic instability occurs early in the progression of cancer. They also demonstrated the remarkable genomic complexity of tumors, with the presence of multiple clones each of which continued to undergo parallel evolution in the primary and metastatic tumors. This is important as it suggests that natural selection does not eliminate all the older, less mutated clones; rather, cancers remain remarkably pluralist societies. In fact, all clones that endure continue to replicate and evolve. Whether these are general findings remains to be clarified, but we can now state that genomic instability plays a major role in the progression of most cancers at some stage in their life.
In addition to the acquired problems mentioned here, inherited factors may contribute to DNA damage and cancer. Thus, mutations or genetic variation at several alleles may either increase susceptibility to DNA-damaging agents such as UV light or compromise the effectiveness of DNA repair. The study of syndromes with inherited defects has greatly contributed to our understanding of how DNA repair is normally executed in mammalian cells. This will be further discussed in this chapter.
Types of Genomic Instability
Where you tend a rose, my lad, a thistle cannot grow.
Frances Hodgson Burnett, The Secret Garden
Most cancers are prone to mutations in DNA as well as more menacing gross chromosomal abnormalities. Genomic instability is not simply the presence of a number of defined mutations, as these are features of all cancers, but rather is the result of shortcomings in DDR that greatly increase the rate at which chromosomes and DNA are damaged and that allow cells with such damage to survive and to replicate. Genomic instability may result from abnormalities across a range of key cellular processes including cell-cycle regulation, DNA damage and repair, aging, and telomere function.
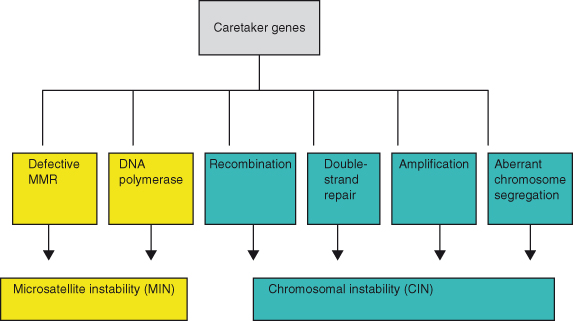
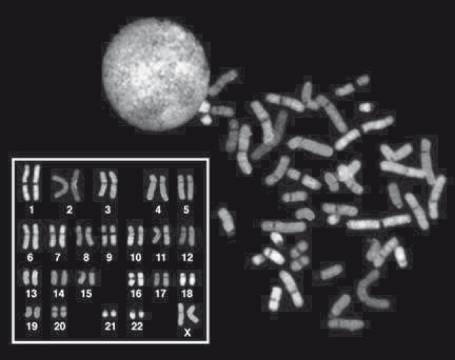
Genomic instability comprises several processes, of which chromosomal instability (CIN) recognized by gross chromosomal abnormalities (deletion and duplication of chromosomes or chromosome parts, re-arrangements, and aneuploidy) and microsatellite instability (MSI) (alterations in the length of short repetitive sequences – microsatellites) associated with a “mutator” phenotype (described in this chapter) have received the most attention (Fig. 10.2). These have already been discussed in the context of colorectal cancers (CRC) in Chapter 3. Aberrant chromosome number (aneuploidy) potentially affects cell populations that undergo multiple cell divisions, including yeast strains, cell lines, and tumor cells, and may result from genome instability. It has been appreciated for almost a century that advanced human cancers invariably contain cells with abnormal numbers of chromosomes, but the role of this in cancer pathogenesis and the likely causes are only recently being elucidated and in some cases remain controversial. The loss or gain of whole chromosomes results primarily from defects of segregation during mitosis, including chromosomal nondisjunction and failure of the mitotic (spindle) checkpoint pathways (Chapters 3 and 4). Normally, unattached kinetochores or those with inappropriate tension during mitosis activate this checkpoint and block mitosis. Mutations in key genes involved in this checkpoint, such as NDC80 and BUB1, may disrupt the mitotic checkpoint and promote CIN. Loss of Rb or p53 may also cause CIN in model systems, possibly through upregulation of Mad2. RB and related pathways appear particularly important for avoiding CIN, as well as for cell-cycle regulation. Chromosome numbers can readily be visualized in skilled hands, by the application of a technique known as spectral karyotyping (SKY). An example of normal human chromosome numbers is shown in Fig. 10.3. Conversely, polyploidy (an increase in chromosome number) in mouse cells is shown in Fig. 10.4. Other types of genomic instability are characterized by an increased rate of small-scale mutations such as MSI. Amplification of individual loci on chromosomes is another form of genetic instability that causes overexpression of proteins, as often occurs for the oncogene c-MYC (Chapter 6).
Figure 10.2 Potential causes of the “mutator” phenotype. Mutations in multiple pathways can result in the mutator phenotype in cancer cells.
A recent study from the laboratory of Titia de Lange has suggested a potential mechanism linking telomere attrition with tetraploidization, a putative precursor to aneuploidy in human cancers. This can be induced by persistent telomere dysfunction if the p53 pathway is defective. Despite undergoing a protracted G2 phase due to activation of both ATR–CHK1 and ATM–CHK2 pathways and missing out on mitosis, cells still degrade the replication inhibitor geminin (by APC/C) and accumulate the origin licensing factor Cdt1. This scenario allows a second S phase and tetraploidy. Worryingly, if telomere protection is restored, such tetraploid cells proliferate.
As you will no doubt have become accustomed to reading by now, there is a lively debate in progress regarding whether genetic instability is a cause or effect of cancer. The CIN hypothesis contends that aneuploidy is the catalyst for transformation, whereas the gene mutation hypothesis asserts that cancer is driven by mutations to proto-oncogenes and tumor suppressor genes, with aneuploidy a side effect of tumorigenesis. The role of point mutations in human cancer is well established, and MSI is widely accepted as causal in tumorigenesis. However, the contribution of massive genomic changes resulting from CIN and aneuploidy is less certain. Aneuploidy is required for sporadic carcinogenesis in mice and may even collaborate with specific gene mutations during tumorigenesis. It has been argued that CIN contributes to cancer initiation because chromosome loss can provide the second hit in loss of a tumor suppressor gene. However, at the same time, CIN is costly for the cell because it destroys the genome and therefore compromises clonal expansion. Interestingly, either CIN or MSI alone may be a sufficient driver of tumorigenesis as, in general, individual tumors manifest one or the other but not both.
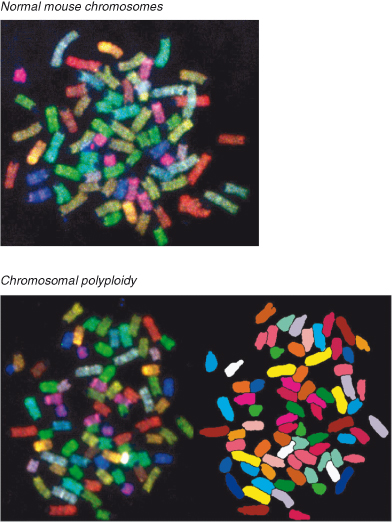
Figure 10.4 Polyploidy in mouse cells as shown by spectral karyotype (SKY). Images
courtesy of Maria Blasco
. SKY provides visualization of all an organism’s chromosomes together, each labeled with a different color, and is readily employed to determine abnormalities in chromosome number. Left: The colors of the different chromosome pairs as seen under microscopy. Right: The polyploid cells have been artificially colored to more easily show the abnormal chromosome numbers – several chromosomes are present in excess numbers (polyploidy).
Mutator Phenotype
The concept of a so-called mutator phenotype of cancers developed from the observation that mutations are rare in normal cells but typical in cancer cells (Fig. 10.2). The mutator phenotype model proposes that the mutation rate in the early stages of tumorigenesis must be greater than the normal spontaneous mutation rate of human somatic cells (1.5 × 10−10 per base pair per cell generation), because this mutation rate is deemed insufficient to produce the multiple mutations identified in many cancers. Note that the mutation rate is much less than the rate of DNA damage, due to the impact of DNA repair. The mutator phenotype implies that genetic instability provides the impetus for accumulation of multiple mutations by the cancer cell. However, only a small number of such mutations may actually confer a growth advantage (and will be selected for), whereas the rest (possibly the majority) have no bearing on the cancer and are merely “baggage” that is not selected for. This differs from the traditional view of cancer progression through clonal evolution driven by mutation and natural selection in so far as the traditional view implies that most mutations will confer some growth advantage and are selected for individually (or in small numbers) – there is little “baggage” in this model. Whatever the answer, most cancers probably develop through a combination of stochastic mutations (and epigenetic factors) as well as those arising through genomic instability (and maybe the epigenetic equivalent, the methylator phenotype, discussed in Chapter 11), which are then subject to natural selection and clonal expansion. However, as discussed in this chapter, particularly in colorectal and breast cancers there is increasing evidence that genetic instability may occur at the transition from premalignant to malignant tumors.
All this interesting debate notwithstanding, the most important issue (and probably one of the most important questions in the whole of cancer biology) is whether the plethora of mutations observed in tumors are all mission-critical or rate-limiting for tumor growth or whether many of the mutations are irrelevant to both the evolution and maintenance of the tumor. In other words, are most mutations in established cancers associated with tumorigenesis in a noncausal way? This has a direct bearing on the design of cancer therapies – new therapeutics should be designed to target cancer-relevant genes or proteins, which must first be distinguished from the rest.
The notion of a “mutator” phenotype is based on seminal studies with one form of a progressive multistage human cancer, a familial colon cancer known as hereditary nonpolyposis colon cancer (HNPCC). In HNPCC, mutation in one allele of the genes involved in DNA mismatch repair is inherited. Loss of the second wild-type allele occurs, frequently promoting the formation of colorectal carcinoma which exhibits MSI. Even sporadic colon cancers often exhibit MSI, and this is an almost ubiquitous finding in HNPCC. Firstly, described in the bacterium Escherichia coli (E. coli), the mismatch repair genes mutS or mutL were implicated in MSI, and subsequently mutations in two homologous human genes in the DNA mismatch repair pathway were found in HNPCC by linkage studies (Chapter 18): MSH2, encoded on chromosome 2, or MLH1, encoded on chromosome 3. These genes may also be inactivated or suppressed by epigenetic factors, though interestingly in human cancers this seems to involve the MLH1 gene (Chapter 11). MSI occurs in several other nonhereditary (sporadic) cancers including, pancreas, colon, and ovary. It is likely that many other genes not necessarily involved in mismatch repair may also contribute to MSI. The human genome comprises vast numbers of microsatellites, but instability also manifests itself in the coding regions of various growth-regulating genes, including apoptosis regulators such as BAX. Thus, MSI may be a “readily measurable” marker of the existence of mutagenic mechanisms, but it must be noted that most sporadic tumors do not display high levels of MSI.
In the case of entosis, aneuploidy may arise by other mechanisms than the failure of cytokinesis due to aberrant mitotic pathways or checkpoints. Sometimes live cells may become internalized by another cell, entosis, resulting in the bizarre appearance of a cell within a cell. Such internalized live cells can persist throughout the cell cycle of the hapless host and cause severe disruption to cytokinesis, thereby resulting in binucleate cells that will father aneuploid cell lineages. Multinucleated cells in human breast cancers sometimes demonstrate such cell-in-cell structures.
Telomere Attrition and Genomic Instability
The cowl makes not the monk.
Walter Scott
Increasing evidence indicates that dysfunctional telomeres likely play a causal role in the process of malignant transformation, in at least a fraction of human cancers, by initiating chromosomal instability. Critical telomeric shortening can lead to telomere “uncapping” and may occur at the earliest recognizable stages of malignant transformation in epithelial tissues. The widespread activation of the telomere-synthesizing enzyme telomerase in human cancers not only confers unlimited replicative potential but also prevents intolerable levels of chromosomal instability. See Chapter 9 for a detailed discussion of this area. There is a considerable overlap between DNA damage responses, particularly to DSBs, and proteins involved in telomere maintenance – see the “Double-strand breaks” section of this chapter.
The DNA Damage Response
The life of every man is a diary in which he means to write one story, and writes another; and his humblest hour is when he compares the volume as it is with what he vowed to make it.
J.M. Barrie
It is all too easy to assume that DNA synthesis is a slipshod affair, littered with errors in spelling and punctuation and successful only because of the imposition of intensive editorial scrutiny and amendment. At this point, I perceive a certain resonance with our own book editor! In fact, errors are remarkably few but are rendered inevitable only because of the enormity of the task.
The human genome, comprising 3 billion base pairs (remember that there are two copies in a somatic cell – so 6 billion bases per cell in total) coding for 30 000–40 000 genes, is under constant assault by endogenous reactive metabolites, therapeutic drugs, and a plethora of environmental mutagens that impact its integrity. Fortunately, the stability of the genome is under continuous surveillance by components of the DDR that will be described in more detail in this section. DNA repair mechanisms have evolved to remove or to tolerate pre-cytotoxic and pre-mutagenic DNA lesions in an error-free (or, in some cases, error-prone) way. Defects in DDR give rise to hypersensitivity to DNA-damaging agents, the accumulation of mutations in the genome, and finally the development of cancer. The importance of DNA repair is illustrated by DNA repair deficiency and genomic instability syndromes (described in this chapter), which are characterized by increased cancer incidence and multiple metabolic alterations. DNA damage, as outlined in Chapter 4, activates two major overlapping networks of proteins with two key apical PI3K-related proteins – the ataxia-telangiectasia mutated (ATM) and ATM- and Rad3-related (ATR) kinases. ATR and ATM, their respective substrates CHK1 and CHK2, as well as the molecules required for recruitment of ATR and ATM to sites of DNA damage, such as histone γH2AX and BRCA1 (see Figs 10.5 and 10.6), are all critical in activating checkpoints (and thereby key proteins involved in these checkpoints as described in Chapters 4 and 7).
Figure 10.5 DNA damage and repair signaling. Signaling mechanisms are activated by DNA damage sensors, ultimately leading to a variety of potential cellular responses discussed earlier.
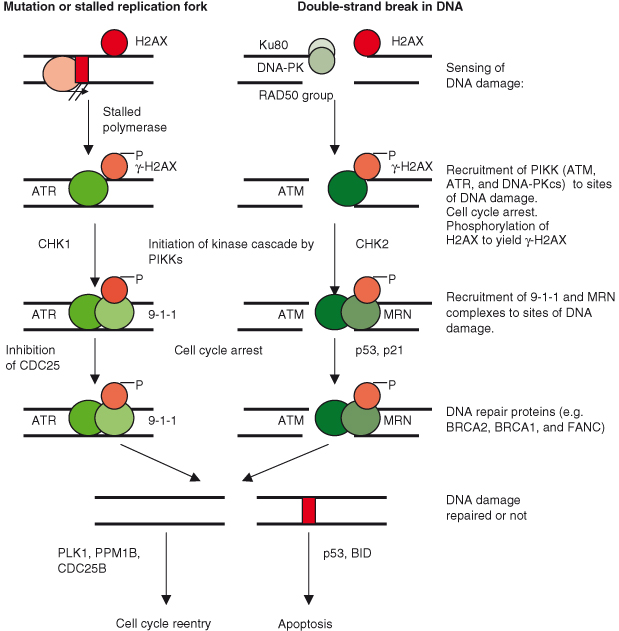
The ATM–CHK2–p53 pathway responds to more substantive damage such as DNA DSBs, whereas the ATR–CHK1 pathway is activated by less extensive DNA damage, including mutations and lesions causing the stalling of polymerases. However, there is considerable overlap and redundancy. A simplified schematic of DNA damage signaling and repair is shown in Fig. 10.5. The effect of such DNA damage response signaling on the cell is to activate cell-cycle checkpoints leading to arrest in the G1 or G2 phase in order for DNA to be repaired before the cell cycle can continue. In certain cases, such as when DNA damage is extensive and/or irreparable, the outcome will be cell death (apoptosis) or irreversible growth arrest (senescence). The development of the concept of cell-cycle checkpoints is attributed to the pioneering work of Lee Hartwell and others starting in the 1960s and has been discussed in Chapter 4. However, given the key role played in avoidance of CIN, the mitotic (spindle) checkpoint will be discussed later in this chapter also. The DNA damage checkpoints coordinate a block in cell proliferation with the DNA repair process. However, once repair is accomplished (if possible), then the checkpoint is “released” and cell division can recommence; Polo-like kinase-1 (PLK1) and Cdc25B are essential components in this reactivation of cell division.
Figure 10.6 DNA damage-induced signaling pathways and repair. (a) Simplified schemata for key DNA damage-induced signaling pathways. Given that p53 functions in the checkpoint response to DSBs, it is not surprising that there is a selective pressure for premalignant tumor cells to mutate and thus inactivate the p53 gene – as is the case for most human cancers. The signaling pathways involved in DNA damage responses are complex and most certainly still incompletely described, but the major proteins involved are detailed here and in more complex detail in (b). Although it is known that various targets of ATM- and ATR-mediated phosphorylation participate in transmitting the DNA damage signal to CHK1/2 proteins, much is still not known. (b) Complex regulation of DNA damage-induced signaling and repair. Signaling involved downstream of different types of DNA damage. To combat recurrent threats of genomic instability, numerous distinct enzyme systems sense DNA damage and coordinate its repair. Part of this coordination involves the activation of signal transduction cascades that target repair proteins, trigger DNA damage-dependent cell cycle checkpoints, and profoundly affect chromatin neighboring a DSB. Ultimately DNA damage results in cell-cycle arrest and attempted repair. If repair is successful, the cell will survive and may re-enter the cell cycle. There is obvious overlap between telomere maintenance and DNA damage response. Proteins directly involved in telomere maintenance and DDR include Ku, DNA-PKcs, RAD51D, PARP-2, WRN, and the MRE11–RAD50–NBS1 complex. The kinases ATM and ATR are key mediators of the DNA damage response and activate downstream effectors such as yH2AX, CHK2, and CHK1. Ultimately the activity of these kinases results in activation of p53 and BID and inhibition of CDC25, leading to growth arrest or apoptosis. Recent studies suggest that P53CSV and/or 14-3-3σ may prevent apoptosis if DNA damage is repairable; and activation of another protein, PPM1D, may allow cell-cycle reentry if DNA damage is repaired. TP53-induced glycolysis and apoptosis regulator (TIGAR) can inhibit glycolysis and production of reactive oxygen species (ROS) levels, both of which contribute to reduction of apoptotic response to p53, potentially also allowing survival in the face of mild or transient stress signals that may be reversed or repaired. Conversely, proteins of the ASPP (ankyrin-repeat-, SH3-domain-, and proline-rich-region-containing proteins) family may preferentially promote the apoptotic activity of p53. 14-3-3σ is a p53 target which may also help maintain G2 arrest. Other 14-3-3 family members may sequester CDC25 and CDC2. Activation of p53 also occurs in response to oncogenic stress (deregulated oncogene activity of e.g. c-Myc), though uniquely in this case p53 is activated by ARF, which displaces the MDM2 negative regulator of p53. In addition to apoptosis, p53 is increasingly shown to also be involved in other forms of cell death. Thus, p53 can induce autophagy, and this is mediated in part at least by damage-regulated autophagy modulator (DRAM), a p53 target gene that encodes a lysosomal protein. DRAM is also essential for p53-mediated apoptosis in response to DNA damage.
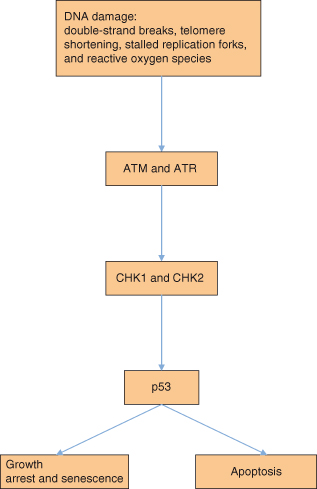
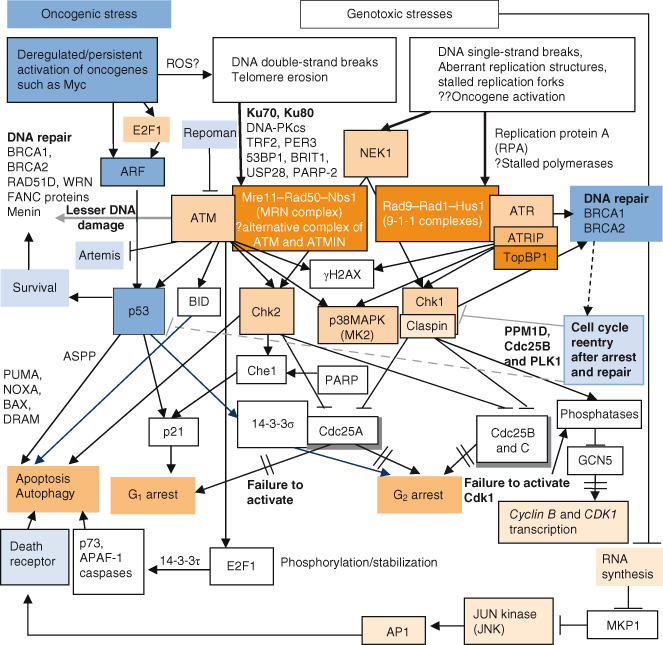
This is a complex and rapidly progressing area of cancer research, and one may be forgiven for approaching a Gordian knot with less trepidation. However, there are rewards awaiting the patient. The basic principles are straightforward and increasingly of very direct relevance to clinical practice. This is illustrated by recent studies which suggest that DDR may be invoked in very early human tumors (even before genomic instability and malignant conversion). Moreover, it is apparent that the DDR is intertwined with general stress responses and with what has been dubbed “oncogenic stress” in particular. Broadly, oncogenic stress may arise due to deregulated activity of a single oncogene, such as c-MYC or RAS, and trigger a failsafe response to eliminate or “hogtie” these potentially dangerous cells – see the “Oncogenic Stress” section of this chapter as well as Chapter 6.
Recent studies highlight the increasing complexity of the DDR, with distinct processes for differing types of damage and also common shared pathways. DNA DSBs are by far the most pernicious type of DNA lesion because, if inefficiently or inaccurately repaired, they can drive genomic instability and cancer. However, DSBs may also result if simpler single-strand lesions are not repaired or replication forks become stalled. Eukaryotic cells have two conserved mechanisms to detect and repair DSBs; homologous recombination (HR) repairs the break using genetic information recovered from the undamaged sister chromatid or chromosomal homologue, whereas nonhomologous end joining (NHEJ) involves the direct ligation of DNA ends. Various proteins are involved in ensuring that chromosomes have been accurately partitioned during mitosis, including those operating in the spindle checkpoint described in this chapter. Mutations in the “caretaker genes” encoding many of these proteins involved in DNA repair and in chromosome segregation and apoptosis have all been implicated in genomic instability. In fact, the caretaker genes represent the third major class of genes mutated in cancer, alongside the oncogenes and tumor suppressor genes.
Sensing DNA Damage
Proofread carefully to see if you any words out.
William Safire
A major focus of recent attention has been the rather tricky issue of how the cell senses DNA damage. As mentioned already, the DNA is normally tightly packed into chromatin by histones – so how is the damaged DNA accessible to the damage-sensing machinery? An expanding body of evidence suggests that sensing may actually take place during the two occasions when DNA is normally stripped of its histone coat and the two strands are separated, namely, during either DNA replication or transcription. It might be argued that as DNA replication occurs during only the S phase of the cell cycle, whereas gene transcription is continuous (except during mitosis, when chromatin is condensed and gene expression is silenced), the latter would be a more suitable occasion to perform such damage sensing. As discussed in Chapter 3, eukaryotic gene transcription involves three different RNA polymerases: RNA polymerase I transcribes ribosomal DNA into ribosomal RNA for ribosome formation, RNA polymerase II transcribes genes into mRNA, and RNA polymerase III synthesizes transfer RNA and small nuclear RNA. Of these, RNA polymerase II reads a much greater proportion of the genome than the others and may moreover recruit DNA-repair proteins to undertake a form of nucleotide excision repair (NER) termed transcription-coupled repair (TCR). This is essential because if RNA polII progress remains stalled because DNA repair is not accomplished, then the cell may trigger other DNA damage sensors, activate the p53 pathway already described in Chapter 7, and even undergo apoptosis.
Potential candidates for the actual DNA damage sensors are discussed under the different types of DNA damage in this chapter, but they include, among other candidates, the so-called 9-1-1 complex (comprising RAD1–RAD9–Hus1) and ATRIP, the MRN complex (MRE11–RAD50–NSB1) and KU80, and the core MMR recognition protein complex, human mutS homolog 2 (hMSH2) and 6 (hMSH6). However, it is still unclear how stalling of DNA or potentially RNA polymerases may activate the DDR. Some proteins involved in transducing the DNA damage signal are described in detail in the “Signaling DNA damage” section.
Signaling DNA Damage
Omnis traductor traditor.
Not surprisingly, mutations that compromise the DDR (e.g. in the ATR–CHK1 or ATM–CHK2–p53 pathway) could propel cell proliferation, survival, increased genomic instability, and thus tumor progression. Examples of such mutations occurring in human cancers are described in this chapter. The labyrinthine signaling pathways involved in the DDR are complex and still incompletely described, but the major proteins involved are detailed in Fig. 10.6. Although it is known that various targets of ATM- and ATR-mediated phosphorylation participate in propagating the DNA damage signal to CHK1 and CHK2 proteins, much is still not known. There is redundancy in the system, and both CHK1 and CHK 2 can inactivate CDC25, thus leading to cell-cycle arrest. Moreover, both CHK2 and ATM may act on the same substrates, providing a potential salvage mechanism if either were mutated. Phosphorylation of the histone variant H2AX by CHK proteins yields γH2AX, which is essential for retention (if not recruitment) of the MRN complex which rapidly accumulates at sites of DNA damage. The MRN complex is a central performer in human genome maintenance. It tethers the broken DNA ends across a strand break and then recruits various repair proteins needed to correctly process the DNA ends. Other important proteins, such as BRCA1, MDC1, and 53BPI (p53 binding protein 1), are also recruited following H2AX phosphorylation. Chromatin modifications are strongly associated with the repair of DSBs, and γH2AX helps prevent aberrant repair of damaged DNA. Mice deficient for both H2AX and p53 rapidly develop tumors. Moreover, even H2AX haploinsufficiency causes genomic instability in normal cells and, on a p53-deficient background, leads to the early onset of lymphoma. H2AX also maps to a cytogenetic region frequently altered in human cancers. Other histone modifications such as acetylation of histone H4 also have an important role in the response to DSBs, suggesting that DNA damage-induced histone modifications are not confined to phosphorylation of H2AX.
Although we know much about the target proteins phosphorylated by ATM, we know little about how ATM is itself phosphorylated and activated, though p53BP1 may be involved. The tumor suppressor protein p53 is a key target of the ATM–CHK2 pathway. Activated p53 induces cell-cycle arrest, which is described in detail in Chapter 7. CHK2 may also activate p21CIP1 independently of p53, contributing to cell-cycle arrest in G1. Given that the p53 pathway functions in the checkpoint response to DSBs, it is not surprising that there is a selective pressure for premalignant tumor cells to mutate and thus inactivate the p53 gene – as is the case for most human cancers.
14-3-3 Proteins
These bind to and influence the activities of a diverse group of molecules involved in signal transduction, cell-cycle regulation, and apoptosis, including p53, RAF, PKC, and BAD. Interactions between 14-3-3 and target proteins are strongly influenced by the phosphorylation state of 14-3-3 and the target protein. The 14-3-3 proteins are remarkably promiscuous in their interactions with target proteins in part due to their ability to associate with both RSXpSXP and RXY–FXpSXP (pS = phosphoserine). In mammalian cells, seven highly conserved isoforms with distinct functions have been identified (14-3-3β, γ, ε, η, σ, τ/θ, and ξ). Several are involved in the G2–M cell-cycle checkpoint; 14-3-3σ, a direct transcriptional target of p53, prevents initiation of mitosis by sequestering cyclin B1–CDK complexes in the cytoplasm, whereas the β and ε isoforms bind Cdc25C. The expression of 14-3-3σ may facilitate DNA damage repair by preventing apoptosis and by arresting cells in G2.14-3-3σ is silenced in a large number of invasive breast cancers and may operate as a tumor suppressor in humans. During a DDR, 14-3-3β and ζ actively promote the retention of activated CHK1 in the nucleus. 14-3-3γ is important for the ability of Chk1 to phosphorylate Cdc25A and target it for degradation.
Repairing DNA Damage
We are the products of editing, rather than of authorship.
George Wald
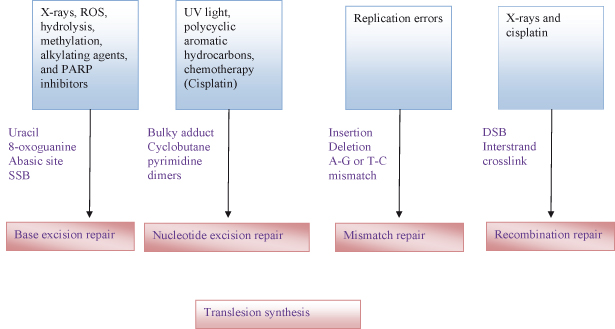
In humans, at least 150 genes encode proteins involved in DNA repair. Various intracellular mechanisms are involved in repairing subtle mistakes made during normal DNA replication or following exposure to DNA-damaging stimuli. Damage due to alkylation can be reversed directly by a chemical process involving genes such as MGMT (no breaking of the DNA backbone is needed). But probably the most important mechanisms are the three excision repair processes – nucleotide excision repair (NER), base excision repair (BER), and mismatch repair (MMR) – all of which may contribute to avoiding MSI and a mutator phenotype. As an illustration of the likely importance of these mechanisms, it has been estimated that on average every somatic cell in humans may need to remove around 10 000–20 000 damaged bases or more every day. Fig. 10.7 summarizes the particular repair mechanisms and the type of damage to which they respond.
Figure 10.7 Various DDR pathways available for the repair of different types of DNA damage. If normal repair fails, tranlesion synthesis may prevent collapsed replication forks and resultant strand breaks.
Fragile Sites
Certain parts of the genome appear particularly vulnerable to damage. Chromosomal fragile sites, which are genomic regions prone to gaps or breaks in metaphase chromosomes during partial replication stress, are particularly vulnerable to DNA breaks and may contribute to chromosomal aberrations in cancer. This view is supported by the frequent identification of tumor suppressors or oncogenes, such as MYC and MET, at fragile sites; the preferential integration of some oncogenic viruses, such as HPV, Hepatitis B, and EBV, at these sites; and the contribution to formation of chromosomal translocations, such as that found in human papillary thyroid carcinoma.
Fragile sites are a normal feature of chromosome architecture, but rare fragile sites can also be inherited. These sites differ in susceptibility to induction by different genotoxic agents and environmental factors. The ATR checkpoint pathway and downstream targets, such as CHK1 and BRCA1, are crucial for fragile site maintenance, suggesting that exposure of single-stranded DNA (ssDNA) during replication stress may activate this checkpoint.
Intriguingly, several putative carcinogens can increase common fragile site breakage in conjunction with agents such as APH and include caffeine, an inhibitor of ATR and ATM; ethanol; cigarette smoking; and pesticides and various other mutagens and chemotherapeutic agents (whose action is enhanced by the addition of caffeine). Recent studies have further supported a role for fragile sites and cancer-specific chromosomal aberrations. Thus, DNA breakage at fragile sites can lead to the formation of RET–PTC rearrangements and also deletions within the FHIT gene similar to those seen in the relevant human cancers.
The Mechanisms of DNA Repair
Various processes contribute to repairing different types of DNA damage and are described in this section.
Single-Stranded DNA Lesions
These are the commonest DNA lesions and may arise by a variety of insults ranging from alterations in a single base through more substantial structural alterations in DNA and chromatin. Unlike DSBs, which are discussed in this chapter, damage is confined to one strand. In general this enables repair by using the normal intact strand as a template, though an error-prone mechanism, known as translesional synthesis (TLS), allows DNA replication to proceed across a damaged template. Several distinct repair processes used to repair single-strand lesions are discussed here.
Base Excision Repair (BER)
Single DNA bases may undergo damage that compromises base pairing by several different chemical reactions, including deamination, oxidation, and alkylation. One noteworthy base substitution results from the incorrect incorporation of adenine opposite to an aberrantly oxidized base, 8-oxoguanine. This results in the conversion of a normal G–C base pair to a T–A one. Another important example is when uracil is incorrectly incorporated into DNA through deamination of cytosine. There are overlaps between BER and repair of SSBs, which can arise directly or as intermediates in BER. In fact, breaks in a single strand of the DNA molecule are repaired using the same enzyme systems that are used in BER.
Two main types of BER are described in humans: a DNA polymerase (Pol) β-dependent pathway and a proliferating cell nuclear antigen (PCNA)-dependent one (also referred to as short- and long-patch repair, respectively). The mechanism of choice may be influenced by various factors including the cell-cycle stage and how resistant the damage is to pol β lyase activity, which is required for short-patch repair. BER (Fig. 10.8
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


