Genetic Disorders
INTRODUCTION
Genetic diseases result from single-gene (mendelian) alterations, chromosomal abnormalities, or multifactorial errors. Over 6,000 such abnormalities have been identified in humans, ranging from mild differences (as in certain hemoglobin abnormalities) to fatal or overwhelmingly disabling conditions, such as trisomy 18 (Edwards’) syndrome and trisomy 13 (Patau’s) syndrome. The risk of single gene disorders is estimated at 1 in 200 births.
Although genetic disorders are determined mainly by genetic makeup, they can also interact with environmental factors. For example, albinism (an inherited inability to generate the protective pigment melanin) greatly increases susceptibility to skin cancer with excessive exposure to sunlight.
GENETIC ANALYSIS
Genetics, the study of heredity, involves analysis of defects in chromosomal reproduction or disease processes that can be passed from one generation to the next. Various tests are used to unravel the effects of altered genes and the patterns of inheritance. As heredity becomes better understood, genetic influences are likely to assume greater importance in health care delivery.
DNA and how it works
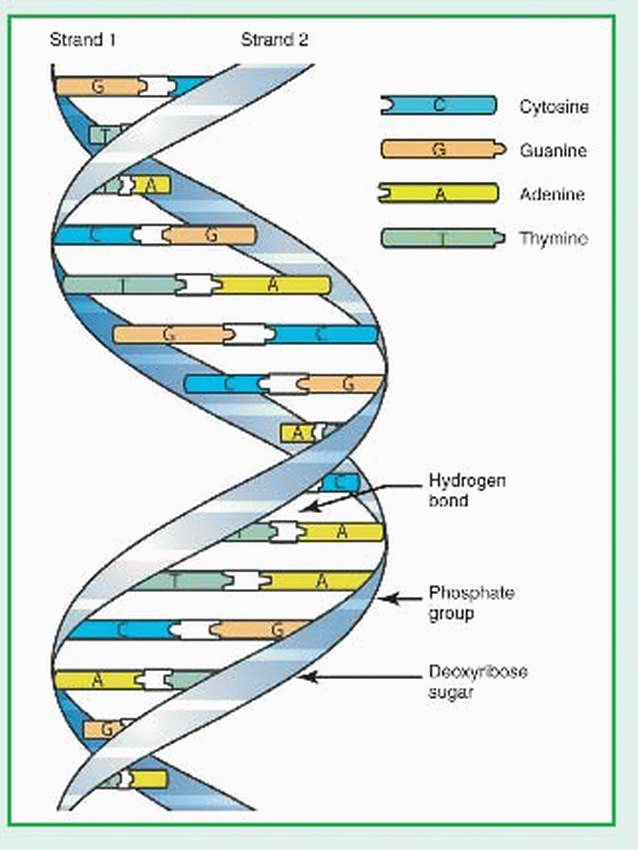
Deoxyribonucleic acid (DNA) is a double-helix polymer (macromolecule) made up of individual units called nucleotides. Each nucleotide is composed of one sugar (deoxyribose), one phosphate, and one nitrogen-containing base—either a purine or a pyrimidine. The purine bases are adenine (A) and guanine (G). The pyrimidine bases are thymine (T) and cytosine (Q). The two polynucleotide chains of each DNA macromolecule are attached by hydrogen bonds between the bases (see illustration). The base pairing is very specific: adenine pairs only with thymine and cytosine pairs only with guanine. The DNA within the human genome (22 different autosomes and two different sex chromosomes) is made up of about 3 billion base pairs.
Genes make up approximately 2% of the human genome. A gene is a segment of DNA that ultimately determines the linear sequence of an amino acid chain. Through a complex process, a gene is transcribed into messenger ribonucleic acid (mRNA), which eventually is translated into an amino acid chain. A sequence of three mRNA nucleotides is called a codon. An mRNA codon can mark the beginning of translation, the end of translation, or a specific amino acid. Our genetic code is made up of 64 different codons, 61 of which actually code for amino acids. The eventual translated strand of amino acids must undergo further modification within the cell before it becomes a functional protein. The Human Genome Project is an international effort that officially began in 1990 and was completed in 2003 with the sequencing of the entire human genome. Emphasis is now on determining how the environment influences gene and protein function. With this information, treatment of genetic-based conditions is occurring at the molecular and cellular levels.
 |
The essential ingredient of heredity is deoxyribonucleic acid (DNA), which makes up genes, basic units of hereditary material that are arranged into threadlike organelles called chromosomes in the cell nucleus. (See DNA and how it works and DNA replication.) Together, these elements contribute to a person’s genotype (gene composition) andphenotype (outward appearance). In humans, each body cell (except ova and sperm) has 46 chromosomes consisting of 22 pairs of autosomes and 1 pair of sex chromosomes. Females have a matched (homologous) pair of X sex chromosomes; males have an unmatched (heterologous) pair of sex chromosomes—an X and a Y. Thus, the normal human chromosome complement is 46,XX in females and 46,XY in
males. The 22 autosomes are all homologous. Each chromosome has a short arm (p) and a long arm (q) joined at the centromere.
males. The 22 autosomes are all homologous. Each chromosome has a short arm (p) and a long arm (q) joined at the centromere.
The position that the gene for a given trait occupies on a chromosome is called a locus. Different loci exist for hair color, blood group, and so on. The number and arrangement of the loci on homologous chromosomes are the same. When the two genes are identical, the individual is homozygous at that locus. When the genes aren’t identical, the individual is heterozygous at that locus. A different form of the same gene that occupies a corresponding locus on a homologous chromosome is called an allele; it determines alternative (and inheritable) forms of the same characteristic. Some alleles control normal trait variation such as hair color; other defective alleles may cause a congenital defect or even induce a spontaneous abortion. Both heterologous (codominant) alleles may express their own effects, or one (the dominant allele) may be expressed and the other (the recessive) suppressed.
A mutation is a permanent change in a DNA sequence. When a mutation occurs in a gene’s DNA sequence it may cause serious, even lethal defects, or it may be relatively benign. Mutations can occur spontaneously or can be caused by exposure to teratogenic agents, such as radiation, drugs, viruses, and synthetic chemicals. Paternal and maternal age also have effects on genetics.
TYPES OF GENETIC DISORDERS
Genetic disorders occur in several different forms. (See Patterns of transmission in genetic disorders, page 986.)
Mendelian or single-gene disorders are inherited in clearly identifiable patterns.
Chromosomal aberrations or abnormalities include structural defects within a chromosome, such as deletion and translocation, plus absence or addition of complete chromosomes.
Multifactorial disorders reflect the interaction of at least two abnormal genes and environmental factors to produce a defect.
SINGLE-GENE DISORDERS
Single-gene disorders may be autosomal (resulting from a single altered gene or a pair of altered genes on one of the 22 pairs of autosomes) or X-linked (resulting from an altered gene on the X chromosome). Single-gene disorders may be further classified as dominant or recessive, depending on whether the altered gene is a dominant or a recessive allele.
DNA replication
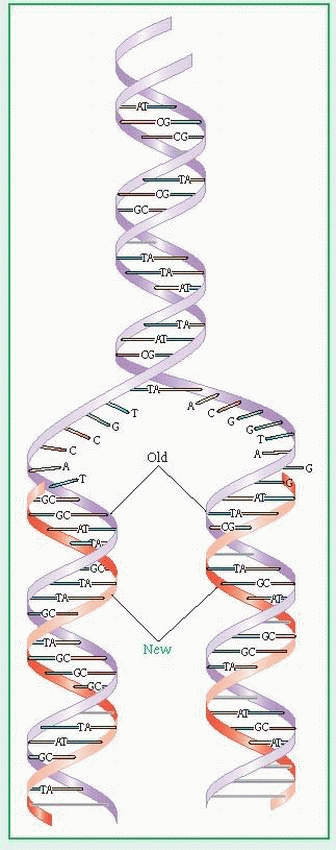
The body grows and replaces all dividing cells other than germ cells (sperm and ova) by mitosis. In mitosis, the cell’s deoxyribonucleic acid (DNA) replicates itself exactly and leads to the creation of a new daughter cell with the identical genetic makeup as the parent cell. Each new cell has a diploid number of chromosomes (in humans, 46).
Germ cells, however, form by meiosis. In meiosis, DNA first replicates. Then, through a complicated process, two cell divisions create four daughter cells (a sperm or ovum) from each parent cell, each of which has a haploid number of chromosomes (in humans, 23).
 |
Patterns of transmission in genetic disorders
Autosomal dominant
Achondroplasia (dwarfism)
Colorectal polyposis
Hereditary hemorrhagic telangiectasia
Huntington’s disease
Hyperlipidemias (some types)
Hypoparathyroidism
Marfan syndrome
Neurofibromatosis (most cases)
Osteogenesis imperfecta (most cases)
Pituitary diabetes insipidus
Polycystic kidney disease
Retinoblastoma
Spherocytosis
Von Willebrand’s disease
Autosomal recessive
Albinism (some cases)
Congenital adrenal hyperplasia
Cretinism
Cystic fibrosis
Cystinuria
Fabry’s disease
Fanconi’s anemia
Galactosemia
Niemann-Pick disease
Osteogenesis imperfecta (some cases)
Phenylketonuria
Polycystic kidney disease
Retinitis pigmentosa (some cases)
Sickle cell anemia
Tay-Sachs disease
Thalassemia (alpha and beta)
Xeroderma pigmentosum (most cases)
X-linked
Duchenne type muscular dystrophy
Fragile X mental retardation
Glucose-6-phosphate dehydrogenase deficiency
Hemophilia (most types)
Pseudohypoparathyroidism
Some immunodeficiencies
Chromosomal
Cri du chat syndrome
Down syndrome (trisomy 21)
Edwards’ syndrome (trisomy 18)
Klinefelter’s syndrome (XXY)
Patau’s syndrome (trisomy 13)
Turner’s syndrome (XO)
Multifactorial
Cleft lip or palate (some cases)
Congenital heart defects (some cases) Diabetes mellitus (some cases)
Mental retardation (some cases)
Neural tube defects
SINGLE-GENE INHERITANCE PATTERNS
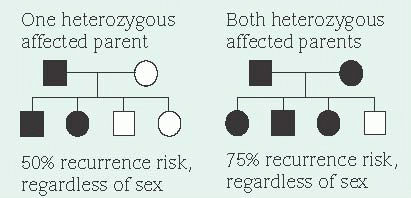
A dominant allele produces its effect in heterozygotes (people who also carry a normal gene for the same trait) because the dominant allele masks the effects of the normal paired gene (autosomal dominant inheritance). Because a person with an autosomal dominant disease is usually a heterozygote and carries one dominant gene as well as one normal gene, his children have a 50% chance of inheriting the dominant gene and the disease. This probability remains the same for each pregnancy. Unaffected people (homozygote for the normal gene) don’t carry the altered gene and therefore can’t transmit it, except as a new mutation. (See Inheritance patterns.)
Sex doesn’t influence transmission of an autosomal dominant allele. However, in some autosomal dominant diseases (such as Huntington’s disease), the severity of symptoms can vary in offspring, depending on which parent transmits the dominant allele. Unless the dominant allele has arisen as a new mutation or is nonpenetrant in an individual, every affected person has an affected parent. Thus, autosomal dominant traits don’t skip generations. However, the severity of symptoms can range from very mild to severe among persons who inherit the allele. This variation of severity is known as expressivity.
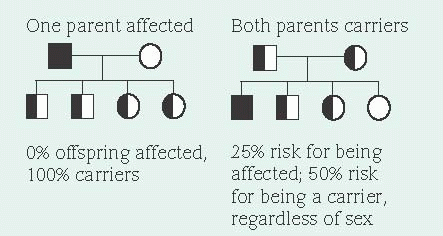
Because a recessive allele can produce a disorder only when paired with another diseasecausing allele (autosomal recessive inheritance), offspring must receive one copy of the disease causing allele from each parent to inherit the recessive trait. A carrier has a diseased gene, but is phenotypically normal. Autosomal recessive disorders affect males and females equally. Because both parents must be heterozygous carriers, autosomal recessive disorders are more common in children of consanguineous parents (blood relatives).
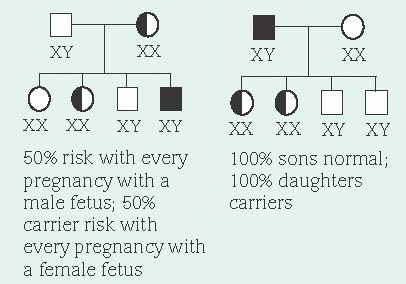
In X-linked recessive inheritance, nearly all affected persons are males because females have two X chromosomes and males have an X and a Y chromosome. There’s very little DNA sequence in common between the X and the Y chromosomes. Therefore, recessive alleles on a male’s X chromosome are expressed. Females who carry a disease-causing recessive allele on only one X
chromosome are usually unaffected because they have two X chromosomes—one with the diseasecausing allele and one with the normal dominant allele. However, every cell in the female, except the oocytes, undergoes a normal process called X inactivation where one X chromosome is turned off. This process is random. Therefore, females who carry only one copy of a disease-causing allele may have mild symptoms if a disproportionate number of cells have the X chromosome with the disease-causing allele turned on and the X chromosome with the normal allele turned off.
chromosome are usually unaffected because they have two X chromosomes—one with the diseasecausing allele and one with the normal dominant allele. However, every cell in the female, except the oocytes, undergoes a normal process called X inactivation where one X chromosome is turned off. This process is random. Therefore, females who carry only one copy of a disease-causing allele may have mild symptoms if a disproportionate number of cells have the X chromosome with the disease-causing allele turned on and the X chromosome with the normal allele turned off.
In X-linked dominant inheritance, which is rare, females are affected to varying degrees. These disorders tend to be lethal in males. A family history of multiple male miscarriages or stillbirths is usually a clue to an X-linked dominant disorder.
Sex does influence transmission of X-linked alleles. An affected male transmits the diseasecausing allele to all of his female offspring, but to none of his male offspring. This is because males receive their X chromosome from their mother. When a sperm containing an X chromosome joins an ovum (which can only have an X chromosome), a female offspring results. When the X chromosome contains a disease-causing recessive allele, female offspring are typically unaffected carriers. When the X chromosome contains a disease-causing dominant allele, all female offspring are affected. Male offspring receive a Y chromosome from their father, therefore not receiving the disease-causing allele.
CHROMOSOMAL ABNORMALITIES
During germ cell formation by meiosis, failure of chromosomes to divide (nondisjunction) results in a germ cell that contains fewer or more than the normal 23 chromosomes. Usually, such abnormal germ cells fail to unite at conception; if fertilization does take place, the embryo is usually miscarried early in the pregnancy. Experts believe that up to 60% of spontaneous abortions at less than 90 days’ gestation result from an abnormal number of fetal chromosomes; offspring with some chromosomal abnormalities are probably never even implanted. Absence of an autosomal chromosome is incompatible with life, but absence of a sex chromosome, as in Turner’s syndrome, is better tolerated. The presence of an extra chromosome (trisomy), as in Down syndrome, commonly produces physical malformation, mental retardation, or both.
Chromosomal disorders may also result from structural changes within chromosomes. For instance, in deletion, loss of part of a chromosome during cell division produces varying effects in the offspring, depending on the type and amount of genetic material lost. An example is velocardiofacial syndrome, in which part of the long arm of chromosome 22 is missing.

In translocation, part of a chromosome attaches itself to another chromosome. If little or no genetic material is lost, the translocation is balanced (symmetrical) and the person displays no effects but may have reproductive problems, such as miscarriage, infertility, or children with malformations, cognitive effects, or both. The person may produce unaffected children, each of whom has a 50% chance of being a balanced translocation carrier, like the parent.
Another abnormality, ring chromosomes, results when a chromosome loses a section of genetic material from each end and the remaining stumps join together to form a ring. The effect varies, depending on the type of genetic material lost and on the specific chromosome that’s involved.
In mosaicism, abnormal chromosomal division in the zygote results in two or more cell lines with different chromosomes. (One cell line may be normal and the other abnormal.) The patient’s phenotype depends on the percentage of normal cells and on the varying effects of the abnormal cell line, which depend on the percentage of abnormal cells in each type of tissue.
Originally, Gregor Mendel’s theories of heredity pointed to the understanding that an individual’s phenotype is the same no matter which parent donates the allele. Now, it’s known that this isn’t always true. When a deletion on 15q11-13 is inherited from the father, the child will have Prader-Willi syndrome (obesity, short structure, hypogonadism). If the deletion is inherited from the mother, the child will have Angelman’s syndrome (mental retardation, no verbal language, seizures). This indicates that different genes are active on chromosome 15 depending on which parent provided the chromosome.
MULTIFACTORIAL DISORDERS
Multifactorial disorders are abnormalities that result from the interaction of at least two inherited abnormal genes and environmental factors. They include common malformations, such as neural tube defects, cleft lip, and cleft palate, as well as disorders that may not appear until later in life such as diabetes mellitus. Such disorders don’t follow the mendelian patterns of inheritance, but the increased incidence of specific birth defects within families suggests familial transmission.
DETECTING GENETIC DISORDERS
Genetic testing and counseling can help prevent genetic disorders and help patients and their families deal with them when they do develop. Genetic diagnosis relies primarily on pedigree (family tree) analysis, karyotype (chromosomal) analysis, and biochemical analysis of blood, urine, or body tissues (including tissues obtained by amniocentesis or chorionic villus sampling [CVS]) to detect abnormal gene products. Neonatal screening for inherited metabolic disorders such as phenylketonuria has become standard, and prompt treatment of such disorders can prevent or minimize their effects.
Simple blood tests can detect carriers of recessive-gene disorders, such as Tay-Sachs disease and sickle cell anemia. DNA testing can detect some autosomal recessive, autosomal dominant, and X-linked recessive disorders. This gives a couple at risk the option of prenatal diagnosis (by amniocentesis or possibly CVS) for some disorders, to detect affected offspring.
A pedigree, a diagram of family relationships and diseases and cause of death of individual family members, helps determine a disorder’s inheritance pattern, including the probability of occurrence. The pedigree chart begins with the patient (proband, or index case) and traces all living and deceased blood relatives in order of their birth, listing:
current age or age at death
health status of all relatives, including miscarriages and stillbirths (and the reasons for them), the site and nature of congenital anomalies, and the presence of mental and growth retardation
relationships (if the patient is a twin, involved in a consanguineous marriage, or divorced)
This information can be obtained from the patient’s memory, autopsy and pathology reports, photographs, and medical records. The pedigree chart is analyzed in relation to the clinical features of the suspected genetic disorder and appropriate laboratory tests or medical records. In one such test, the karyotype, white blood cells obtained from a venous blood sample are grown in a special culture until a specific stage of mitosis, when chromosomes are most easily seen with a microscope. Then the cells are broken open and stained to show specific bands on the chromosomes. Staining techniques can be varied to help identify each chromosome and the bands it contains.
Amniocentesis, needle aspiration of amniotic fluid after transabdominal puncture of the uterus under ultrasound guidance, can now detect more than 600 genetic disorders before birth by:
karyotyping cultured amniocytes
using fluorescent in situ hybridization on cultured amniocytes to detect submicroscopic chromosome deletions, duplications, or translocations
measuring enzyme levels or activity on cultured amniocytes
performing DNA mutation or linkage analysis on cultured amniocytes
measuring proteins (e.g., alpha-fetoprotein) or biochemical substrates in amniotic fluid.
Amniocentesis allows parents to make informed decisions for a pregnancy before the birth of a child with a genetic disorder. It’s recommended to patients with:
maternal age over 34 at delivery
family history of chromosomal abnormalities
history of previous children or a first- or second-degree relative with an open or closed neural tube defect or scoliosis with spinal dysraphism
history of multiple reproductive losses
parents who are known carriers of a genetic disorder that’s detectable by biochemical or DNA testing
The benefits of amniocentesis must be carefully weighed against the potential complications, which may include amniotic fluid leakage, miscarriage, and (rarely) infection. The risk of complications is estimated to be 0.5% (1 in 200) or less. Genetic counseling is done to help the patient and family weigh the risks against the benefits so they can make an informed decision about testing. For some patients, CVS, another method of collecting genetic information from the fetus, may be an alternative. Using a vaginal or abdominal approach, with ultrasound as a guide, the physician collects a small amount of chorionic tissue, which is then analyzed in much the same way as the amniocentesis. Biochemical tests that are typically done on amniotic fluid can’t be done on CVS tissue.
Researchers are currently studying new methods of earlier prenatal diagnosis, such as amniocyte filtration and karyotyping of fetal cells in maternal blood.
Prenatal diagnosis may be considered even when a couple would continue an affected pregnancy because knowing the diagnosis ahead of time can help the health care team provide the optimal timing and method of delivery, thus improving the neonate’s outcome. It also gives the couple more time to look into financial, educational, and psychological support services to prepare for the birth of a child with special needs.
Health care providers may recommend genetic testing for selected children and adolescents; it’s indicated for prevention, decreasing the need for surveillance, and examining treatment availability.
HELPING THE FAMILY COPE
Genetic counseling helps a family to understand its risk for a particular genetic disorder and to cope with the disorder if that risk becomes reality. Counseling sessions make it easier for the family to comprehend:
medical facts (diagnosis, prognosis, treatment)
how heredity works (risks to other relatives)
options for dealing with the problem
consequences of their decision
Psychological support to relieve stress and improve the child’s or parents’ self-concept is as important as obtaining the correct information. The birth of a child with a genetic defect may provoke parental feelings of guilt, anxiety, isolation, insecurity, and helplessness and may place undue stress on all members of the family. Family members may experience a period of shock and denial, followed by grief and mourning. Following acceptance of the diagnosis, parents may continue to experience periods of denial, guilt, and chronic sorrow, a phenomenon that manifests as episodes of sadness, particularly around developmental milestones. These feelings are a normal response to having a child with a disability.
When testing confirms a genetic disorder, here’s how you can provide psychological support:
Refer the family for genetic counseling. The following organizations can be contacted to obtain a list of qualified health professionals throughout the United States who can provide genetic evaluation, diagnosis, counseling, and management services:
American College of Medical Genetics
International Society of Nurses in Genetics
March of Dimes Birth Defects Foundation
National Society of Genetic Counselors
Find out the services that are offered by the counseling center so you can tell the family what to expect.
Provide the genetic professional with pertinent medical records and information about the family’s special concerns, such as religious beliefs and social preferences.
After counseling sessions, make sure family members understand the new information presented to them, and reinforce the information provided. Communicate with them in an unhurried and nontechnical manner, and make sure your facts are accurate.
Inform the family about community resources, agencies, and available support groups to help them deal with genetic disorders. If they’re interested, help them get in touch with the families of other patients with the same disorder. The Alliance of Genetic Support Groups or the National Organization for Rare Disorders are valuable resources for locating support groups and parent networking opportunities.
Coordinate the assistance needed from other members of the health care team, such as physicians, psychologists, and social workers.
Recognize the parents’ stresses in caring for their child, and allow them to express their feelings.
AUTOSOMAL DOMINANT INHERITANCE
Neurofibromatosis
Neurofibromatosis is a group of inherited developmental disorders of the nervous system, muscles, bones, and skin that causes formation of multiple, pedunculated, soft tumors (neurofibromas), and café-au-lait spots. The most common types are NF-1 (von Recklinghausen disease) and NF-2 (bilateral acoustic neurofibromatosis). About 80,000 Americans are known to have neurofibromatosis; in many others, the disorder is overlooked because symptoms are mild. The prognosis varies; however, spinal or intracranial tumors can shorten the patient’s life span.
CAUSES AND INCIDENCE
NF-1 is an autosomal dominant disorder of chromosome 17 that occurs in about 1 in 3,000 births. About 50% of affected families have a negative family history; in many of these families, the father is older, suggesting that advanced paternal age may influence the NF-1 mutation. NF-2 is an autosomal dominant disorder of chromosome 22; however, many patients have a negative family history.
SIGNS AND SYMPTOMS
Signs and symptoms of NF-1 vary greatly from one family to another and within members of the same family. A patient who initially seems to have mild symptoms may develop more severe problems later.
An infant with this form may present with only café-au-lait spots or may also have congenital glaucoma, plexiform neurofibromas, or pseudoarthrosis. About 90% of patients have Lisch nodules on the iris; as many as 15% develop optic pathway gliomas, which may cause a significant loss of vision.
Cutaneous and other neurofibromas may begin to develop or become more prominent at puberty; pregnancy may exacerbate tumor growth. Some tumors become malignant; about 8% of patients develop neurofibrosarcoma (cancer of the nerve sheath). Other less-specific features may include other types of tumors (such as meningiomas), short stature, seizures, speech and learning disabilities, mental retardation (occasionally), and abnormalities of the cerebral, GI, and renal arteries.
The first sign of NF-2 is usually a central nervous system tumor, such as a spinal or intracranial meningioma, an acoustic neuroma, and occasionally a schwannoma or spinal astrocytoma. Cutaneous neurofibromas may be less conspicuous in this form, and café-au-lait spots may be minimal or even absent. Learning disabilities and other less-specific features characteristic of NF-1 aren’t typically seen in NF-2.
DIAGNOSIS
Diagnosis rests on typical clinical findings, especially neurofibromas and café-au-lait spots. Diagnostic criteria for NF-1 include two or more of the following:
first-degree relative (parent, sibling, child) with known NF-1
six or more café-au-lait spots over 5 mm in diameter in a prepubertal patient and 15 mm or more in a postpubertal patient
freckling in the axillary or inguinal region
optic pathway glioma
two or more Lisch nodules on the iris
osseous lesion, such as sphenoid dysplasia or thinning long-bone cortex
Diagnostic criteria for NF-2 include a firstdegree relative with NF-2 and two of the following:
neurofibroma, meningioma, schwannoma, glioma, juvenile posterior subcapsular lenticular opacity
unilateral eighth cranial nerve mass
bilateral eighth cranial nerve masses seen on magnetic resonance imaging (MRI) or computed tomography (CT) scan
X-rays, MRI, and CT scan may be indicated to determine the presence of widening internal auditory meatus and intervertebral foramen. An eye examination to look for Lisch nodules should be done on patients suspected of having NF-1. Myelography may be used to identify spinal cord tumors, and lumbar puncture with cerebrospinal fluid analysis will reveal elevated protein concentration in the presence of spinal neurofibromas and acoustic tumors. Deoxyribonucleic acid analysis and prenatal diagnosis may also be done in some families.
TREATMENT
Neurofibromatosis has no specific treatment. Management consists of surgical removal of intracerebral or intraspinal tumors (when possible) and correction of kyphoscoliosis. Tumors that cause pain and loss of function are removed on an individual basis. If the entire tumor can’t be removed during surgery,
radiation may also be used to help relieve symptoms.
radiation may also be used to help relieve symptoms.
Tumors that grow rapidly should be removed promptly because they may become malignant.
Cosmetic surgery for disfiguring or disabling growths may be done; however, regrowth is likely. Special schooling for individuals with learning disorders or attention deficit hyperactivity disorder may be required. Annual eye exams should be performed.
Researchers are currently investigating experimental treatments for severe tumors.
SPECIAL CONSIDERATIONS
Disfigurement may cause overwhelming embarrassment and social regression. By showing acceptance, you can help the patient adjust to his condition.
Advise the patient to choose attractive clothing that covers nodules; suggest special cosmetics to cover skin lesions.
Refer the patient for genetic counseling to discuss the 50% risk of transmitting this disorder to offspring. Recommend contacting the National Neurofibromatosis Foundation.
Osteogenesis imperfecta
Osteogenesis imperfecta (brittle bones) is a hereditary disease of bones and connective tissue that may cause varying degrees of skeletal fragility, thin skin, blue sclerae, poor teeth, hypermobility of joints, and progressive deafness. This disease occurs in many forms. In the rare congenital form, fractures are present at birth. This form is usually fatal within the first few days or weeks of life. In the late-appearing form, the child appears normal at birth but develops recurring fractures (mostly of the extremities) after the first year of life.
CAUSES AND INCIDENCE
Osteogenesis imperfecta can result from autosomal dominant inheritance of a defect in the amount of Type I collagen, an important part of the bone matrix. Clinical signs may result from defective osteoblastic activity and a defect of mesenchymal collagen (embryonic connective tissue) and its derivatives (sclerae, bones, and ligaments). The reticulum fails to differentiate into mature collagen or causes abnormal collagen development, leading to immature and coarse bone formation. Cortical bone thinning also occurs.
Type I, the most common form of osteogenesis imperfecta, occurs in about 1 in 30,000 live births. Both types I and IV are thought to be inherited as an autosomal dominant trait. Types II and III are believed to be inherited as an autosomal recessive trait.
COMPLICATIONS
Deafness (caused by otosclerosis)
Scoliosis
Multiple deformities
Short stature
SIGNS AND SYMPTOMS
Clinical severity varies, depending on the type. In type I, fractures characteristically occur from minimal trauma. The sclerae are a deep blue-black color, and the teeth may be yellow or even grayish blue from opalescent dentin. Patients with dental abnormalities are shorter and have more fractures at birth, more frequent fractures, and more severe skeletal deformities than type I patients with normal teeth.
Bowing of the lower limbs is common in this type, as is kyphosis in adults. About 40% of all adults with type I have severely impaired hearing, and virtually all adults have some degree of hearing impairment by age 50. The number of fractures may spontaneously decrease in adolescence.
Type II is characterized by intrauterine fractures due to extreme bone fragility, leading to intrauterine or early infant death. Death usually results from complications of bone fragility, heart failure, pulmonary hypertension, or respiratory failure. Therapeutic intervention doesn’t usually increase survival.
Type III is generally nonlethal. Fractures are usually present at birth and occur frequently in childhood; they typically lead to progressive skeletal deformity and, eventually, impaired mobility. Patients have a poor growth rate; most fall below the third percentile in height for their age. Their sclerae are usually normal or light blue, and their teeth aren’t usually opalescent.
Type IV is characterized by osteoporosis, which leads to increased bone fragility. The sclerae may be light blue at birth but appear normal in adolescents and adults. Bowed limbs may be present at birth, but only 25% of patients have fractures at birth. The number of fractures may decrease spontaneously at puberty, but the majority of patients are short. A few have a skull deformity.
DIAGNOSIS
Family history and characteristic features, such as blue sclerae or deafness, establish the diagnosis. Whenever possible, collagen biochemical
studies of cultured skin fibroblasts should be performed. Prenatal diagnosis may be available for certain families with an identified mutation. Prenatal ultrasound performed as early as 16 weeks’ gestation may show evidence of severe osteogenesis imperfecta. X-rays showing evidence of multiple old fractures and skeletal deformities and a skull X-ray showing wide sutures with small, irregularly shaped islands of bone (wormian bones) between them support the diagnosis. These findings can help differentiate osteogenesis imperfecta from child abuse or from other disorders such as juvenile idiopathic osteoporosis.
studies of cultured skin fibroblasts should be performed. Prenatal diagnosis may be available for certain families with an identified mutation. Prenatal ultrasound performed as early as 16 weeks’ gestation may show evidence of severe osteogenesis imperfecta. X-rays showing evidence of multiple old fractures and skeletal deformities and a skull X-ray showing wide sutures with small, irregularly shaped islands of bone (wormian bones) between them support the diagnosis. These findings can help differentiate osteogenesis imperfecta from child abuse or from other disorders such as juvenile idiopathic osteoporosis.
In a family with a history of type II osteogenesis imperfecta, diagnostic serial ultrasound should be considered for future pregnancies to detect limb shortening, in utero fractures, and polyhydramnios.
TREATMENT
Treatment aims to prevent deformities by traction, immobilization, or both and to aid normal development and rehabilitation. Fractures must be repaired quickly to avoid deformities. Surgical procedures such as inserting metal rods through bones can help strengthen bones and prevent deformity. The use of bisphosphonates in children with osteogenesis imperfecta is being researched, as are growth hormone and gene therapies. Starting bisphosphonates after diagnosis at birth decreases fracture rates and helps spur growth. Research is also being done on the benefits of bone marrow transplantation and increasing density in trabecular bone formation. Supportive measures include:
checking the patient’s circulatory, motor, and sensory abilities
encouraging the patient to walk when possible (Children with osteogenesis imperfecta develop a fear of walking.)
teaching preventive measures, such as avoiding contact sports or strenuous activities or wearing knee pads, helmets, or other protective devices when engaging in sports
assessing for and treating scoliosis, a common complication
promoting preventive dental care and repair of dental caries
SPECIAL CONSIDERATIONS
Educate the family about the disorder. Teach the parents and child how to recognize fractures and how to correctly splint them. Also teach the parents how to protect the child during diapering, dressing, and other activities of daily living.
Advise the parents to encourage their child to develop interests that don’t require strenuous physical activity and to develop his fine motor skills. These actions will promote the child’s self-esteem.
Advise the parents that physical and rehabilitation therapy is beneficial. Swimming is an excellent conditioning exercise.
Teach the child to assume some responsibility for precautions during physical activity to help foster his independence.
Stress the importance of good nutrition to heal bones and optimize muscle strength.
Refer the parents and child for genetic counseling to assess the recurrence risk.
Administer analgesics, as ordered, to relieve pain from frequent fractures, a hallmark of this disease.
Monitor dental and hearing needs. Stress the need for regular dental care and immunizations.
Instruct the parents to provide a medical identification bracelet for the child.
Marfan syndrome
Marfan syndrome is a rare, predominantly inherited, degenerative, generalized disease of the connective tissue that causes ocular, skeletal, and cardiovascular anomalies. It probably results from elastin and collagen abnormalities. Death is usually attributed to cardiovascular complications and may occur any time from early infancy to adulthood, depending on the severity of the symptoms. Marfan syndrome affects males and females equally.
CAUSES AND INCIDENCE
Marfan syndrome is inherited as an autosomal dominant trait of chromosome 15. It’s caused by mutations in the fibrillin-1 gene, producing changes in elastic tissues, especially of the aorta, eye, and skin. Mutations of the fibrillin-1 gene also cause overgrowth of long bones. In 85% of patients with this disease, the family history confirms Marfan syndrome in one parent as well. In the remaining 15%, a negative family history suggests a fresh mutation, possibly from advanced paternal age.
SIGNS AND SYMPTOMS
The most common signs and symptoms of this disorder are skeletal abnormalities, particularly excessively long tubular bones and an arm span that exceeds the patient’s height. The patient is usually taller than average for his family (in the 95th percentile for his age), with the upper half of his body shorter than average and the lower half, longer. His fingers are long and slender (arachnodactyly). Weakness of ligaments, tendons, and joint capsules results in joints that are loose, hyperextensible, and habitually dislocated. Excessive growth of the rib bones gives rise to chest deformities such as pectus excavatum (funnel chest).
Eye problems are also common; 75% of patients have crystalline lens displacement (ectopia lentis), the ocular hallmark of Marfan syndrome. Quivering of the iris with eye movement (iridodonesis) typically suggests this disorder. Most patients are severely myopic, many have retinal detachment, and some have glaucoma.
The most serious complications occur in the cardiovascular system and include weakness of the aortic media, which leads to progressive dilation or dissecting aneurysm of the ascending aorta. Such dilation appears first in the coronary sinuses and is commonly preceded by aortic insufficiency. Less-common cardiovascular complications include mitral valve prolapse and endocarditis.
Other associated problems include sparsity of subcutaneous fat, frequent hernias, cystic lung disease, recurrent spontaneous pneumothorax, and scoliosis or kyphosis.
DIAGNOSIS
Because no specific test confirms Marfan syndrome, diagnosis is based on typical clinical features (particularly skeletal deformities and ectopia lentis) and a history of the disease in close relatives. Useful supplementary procedures, though not definitive for diagnosis, include X-rays for skeletal abnormalities and an echocardiogram to detect aortic root dilation. Eye examination with slit-lamp may be used to diagnose a dislocated lens.
TREATMENT
Attempts to stop the degenerative process have met with little success. Therefore, treatment of Marfan syndrome is basically aimed at relieving symptoms—for example, surgical repair of aneurysms and ocular deformities. In young patients with early dilation of the aorta, prompt treatment with beta-adrenergic blockers may decrease ventricular ejection and protect the aorta; extreme dilation requires surgical replacement of the aorta and the aortic valve. Steroids and sex hormones have been successful (especially in girls) in inducing precocious puberty and early epiphyseal closure to prevent abnormal adult height. Genetic counseling is important, particularly because pregnancy and resultant increased cardiovascular workload can produce aortic rupture.
SPECIAL CONSIDERATIONS
High school and college athletes (particularly basketball players) who fit the criteria for Marfan syndrome should undergo a careful clinical and cardiac examination before being allowed to play, to avoid sudden death due to dissecting aortic aneurysm or other cardiac complications.
Provide the patient with supportive care, as appropriate for his clinical status.
Educate the patient and his family about the course of the disease and its potential complications.
Stress the need for frequent checkups to detect and treat degenerative changes early.
Emphasize the importance of taking prescribed medications and of avoiding contact sports and isometric exercise.
To encourage normal adolescent development, advise the parents to avoid unrealistic expectations for their child simply because he’s tall and looks older than his years.
Refer the patient and family to the National Marfan Foundation for additional information.
Stickler’s syndrome
Stickler’s syndrome also called (arthro-ophthalmopathy) is an autosomal dominant chondrodysplasia caused by structural defects in collagen. It’s characterized by ocular, skeletal, auditory, and craniofacial abnormalities. Expression of clinical features is highly variable from one individual to another.
CAUSES AND INCIDENCE
At least 19 different types of collagen have been identified. Collagen is an essential component of connective tissues. The collagen defect in most families with Stickler’s syndrome is caused by a mutation in the type II collagen gene (COL2A1) located on chromosome 12q13. Other families demonstrate linkage to the COL11A2 gene located on chromosome 6p21.3 and others to the COL11A1 gene located on
chromosome 1p21. COL2A1 and COL11A1 are expressed in the hyaline cartilage, vitreous, intervertebral disk, and inner ear. COL11A2 isn’t expressed in the vitreous. Genetic studies have identified a few families who carry the clinical diagnosis of Stickler’s syndrome yet don’t demonstrate linkage to any of the three aforementioned collagen genes, suggesting further genetic heterogeneity (a pattern of traits caused by genetic factors in some cases and nongenetic factors in others).
chromosome 1p21. COL2A1 and COL11A1 are expressed in the hyaline cartilage, vitreous, intervertebral disk, and inner ear. COL11A2 isn’t expressed in the vitreous. Genetic studies have identified a few families who carry the clinical diagnosis of Stickler’s syndrome yet don’t demonstrate linkage to any of the three aforementioned collagen genes, suggesting further genetic heterogeneity (a pattern of traits caused by genetic factors in some cases and nongenetic factors in others).
About 1 in 10,000 people is affected with Stickler’s syndrome. However, this incidence rate is considered conservative because persons with very mild symptoms may never be diagnosed with the syndrome.
COMPLICATIONS
Glaucoma
Retinal detachment
Deafness
Osteoarthritis
Ear infections
Difficulty breathing or feeding
SIGNS AND SYMPTOMS
The clinical phenotype can consist of ocular, auditory, craniofacial, and skeletal abnormalities. The number of organ systems involved and the specific phenotypic features expressed can vary significantly between affected family members and, in particular, between unrelated affected persons.
Ocular symptoms, particularly high myopia, are common in persons with Stickler’s syndrome, with the exception of those who have COL11A2 mutations. Vitreal abnormalities are considered a hallmark of Stickler’s syndrome, although the abnormalities in the vitreous differ in persons with a COL2A1 mutation from those with a COL11A1 mutation. Retinal detachment resulting in blindness is the most serious ocular complication.
The vitreoretinal degeneration that leads to retinal detachment is much more common in persons with a COL2A1 mutation. Persons with Stickler’s syndrome can also have congenital cataracts and develop glaucoma. Ocular symptoms are typically absent in persons with linkage to COL11A2.
Auditory symptoms include conductive hearing loss secondary to Eustachian tube dysfunction in children with cleft palate or collagen defects in the inner ear apparatus. Sensorineural hearing loss has an earlier onset and tends to be more progressive in persons with a COL11A1 mutation.
Craniofacial features may include micrognathia (small lower jaw) and a flattened midface and nasal bridge. Micrognathia may be associated with some degree of cleft palate (bifid uvula to complete cleft of the palate). Micrognathia associated with glossoptosis places neonates and infants with Stickler’s syndrome at significant risk for episodic obstructive apnea during feeding and when lying flat.
Skeletal symptoms can include joint hypermobility in young children, spondyloepiphyseal dysplasia, and, later, degenerative arthropathy during early adult years. Also related to the collagen defect, scoliosis and mitral valve prolapse can develop in some persons with Stickler’s syndrome.
DIAGNOSIS
Diagnosis is based on the recognition of clinical features consistent with Stickler’s syndrome. The number of genes involved in Stickler’s syndrome and the complexity of sequencing large genes currently preclude the clinical utility of routine genetic testing for diagnostic purposes. Therefore, diagnosis must rely on the dysmorphology skills of the practitioner performing the physical examination.
Stickler’s syndrome should be considered in neonates with the triad of micrognathia, cleft of the soft palate, and glossoptosis. This triad of features is known as Pierre Robin syndrome.
Severe myopia in infants and young children should be considered a possible symptom of Stickler’s syndrome. The diagnosis should also be considered in persons with a family history of cleft palate and significant myopia, deafness, or spondyloepiphyseal dysplasia. A slit-lamp examination of the vitreous is necessary to determine the presence of vitreal abnormalities that are considered pathognomonic of Stickler’s syndrome.
TREATMENT
Beginning with the first 6 months after birth, annual ophthalmology evaluation must be obtained to assess for myopia, vitreal abnormalities, and retinal degeneration and detachment. Persons who experience floaters or shadows in their vision require immediate assessment.
Persons with vitreoretinal degeneration need to avoid contact sports and other physical activity that can jar and detach the retina.
A brain stem auditory evoked response evaluation should be done during the first month after birth. The schedule for regular follow-up needs to be determined based on the results
of the initial evaluation, frequency of otitis media episodes, and progress in language development.
of the initial evaluation, frequency of otitis media episodes, and progress in language development.
Early use of corrective lenses and hearing aids is recommended to enable developmental progression at the infant or young child’s full potential.
Depending on the infant’s weight and health, surgical correction of the cleft palate can occur around age 9 months. With few exceptions, surgical closure of the palate should occur before age 2 to maximize speech and language development.
SPECIAL CONSIDERATIONS
To assess for obstructive apnea, neonates with the Pierre Robin syndrome triad need oxygen and carbon dioxide measurements while lying in different positions and while feeding.
Neonates and infants with cleft palate may need a specialized cleft palate nurser to obtain adequate caloric intake.
Screening for cardiac valvular disease should be considered in the older child and adult with Stickler’s syndrome.
Genetic counseling by a person trained in genetics should be offered to adults with Stickler’s syndrome.
Referral to appropriate support groups or networking groups should be offered to help with coping skills and to provide anticipatory guidance related to day-to-day needs of a person with Stickler’s syndrome.
AUTOSOMAL RECESSIVE INHERITANCE
Cystic fibrosis
Cystic fibrosis is a generalized dysfunction of the exocrine glands that affects multiple organ systems. Transmitted as an autosomal recessive trait, it’s the most common fatal genetic disease in White children.
Cystic fibrosis is a chronic disease. With improvements in treatment over the past decade, the average life expectancy has risen from age 16 to age 40 and older.
CAUSES AND INCIDENCE
The gene responsible for cystic fibrosis (located on chromosome 7) encodes a protein that involves chloride transport across epithelial membranes; more than 100 specific mutations of the gene are known. (See Cystic fibrosis transmission risk, page 996.) The immediate causes of symptoms in cystic fibrosis are increased viscosity of bronchial, pancreatic, and other mucous gland secretions and consequent obstruction of glandular ducts. Cystic fibrosis accounts for almost all cases of pancreatic enzyme deficiency in children.
In the United States, the incidence of cystic fibrosis is highest in Whites of northern European ancestry (1 in 2,000 live births) and lowest in Blacks (1 in 17,000 live births), Native Americans, and people of Asian ancestry. The disease occurs equally in both sexes.
COMPLICATIONS
Bronchiectasis
Pneumonia
Atelectasis
Hemoptysis
Dehydration
Distal intestinal obstruction syndrome
Malnutrition
Nasal polyps
Gastroesophageal reflux
Rectal prolapse
Cor pulmonale
Diabetes
Pancreatitis
Cholecystitis
SIGNS AND SYMPTOMS
The clinical effects of cystic fibrosis may become apparent soon after birth or may take years to develop. They include major aberrations in sweat gland, respiratory, and GI function. Sweat gland dysfunction is the most consistent abnormality. Increased concentrations of sodium and chloride in the sweat lead to hyponatremia and hypochloremia and can eventually induce fatal shock and arrhythmias, especially in hot weather.
Respiratory symptoms reflect obstructive changes in the lungs: wheezy respirations; a dry, nonproductive paroxysmal cough; dyspnea; and tachypnea. These changes stem from thick, tenacious secretions in the bronchioles and alveoli and eventually lead to severe atelectasis and emphysema. Children with cystic fibrosis display a barrel chest, cyanosis, and clubbing of the fingers and toes. They suffer recurring bronchitis and pneumonia as well as associated nasal polyps and sinusitis. Death typically results from pneumonia, emphysema, or atelectasis.
The GI effects of cystic fibrosis occur mainly in the intestines, pancreas, and liver. One early
symptom is meconium ileus; the neonate with cystic fibrosis doesn’t excrete meconium, a dark green mucilaginous material found in the intestine at birth. He develops symptoms of intestinal obstruction, such as abdominal distention, vomiting, constipation, dehydration, and electrolyte imbalance. As the child gets older, obstruction of the pancreatic ducts and resulting deficiency of trypsin, amylase, and lipase prevent the conversion and absorption of fat and protein in the GI tract. The undigested food is then excreted in frequent, bulky, foul-smelling, pale stools with a high fat content. This malabsorption induces poor weight gain, poor growth, ravenous appetite, distended abdomen, thin extremities, and sallow skin with poor turgor. The inability to absorb fats results in a deficiency of fat-soluble vitamins (A, D, E, and K), leading to clotting problems, retarded bone growth, and delayed sexual development. Males may experience azoospermia and sterility; females may experience secondary amenorrhea but can reproduce. A common complication in infants and children is rectal prolapse secondary to malnutrition and wasting of perirectal supporting tissues.
symptom is meconium ileus; the neonate with cystic fibrosis doesn’t excrete meconium, a dark green mucilaginous material found in the intestine at birth. He develops symptoms of intestinal obstruction, such as abdominal distention, vomiting, constipation, dehydration, and electrolyte imbalance. As the child gets older, obstruction of the pancreatic ducts and resulting deficiency of trypsin, amylase, and lipase prevent the conversion and absorption of fat and protein in the GI tract. The undigested food is then excreted in frequent, bulky, foul-smelling, pale stools with a high fat content. This malabsorption induces poor weight gain, poor growth, ravenous appetite, distended abdomen, thin extremities, and sallow skin with poor turgor. The inability to absorb fats results in a deficiency of fat-soluble vitamins (A, D, E, and K), leading to clotting problems, retarded bone growth, and delayed sexual development. Males may experience azoospermia and sterility; females may experience secondary amenorrhea but can reproduce. A common complication in infants and children is rectal prolapse secondary to malnutrition and wasting of perirectal supporting tissues.
Cystic fibrosis transmission risk
The chance that a relative of a person with cystic fibrosis or a person with no family history will carry the cystic fibrosis gene appears in the chart below.
Relative of affected person | Carrier chance |
Brother or sister | 2 in 3 (67%) |
Niece or nephew | 1 in 2 (50%) |
Aunt or uncle | 1 in 3 (33%) |
First cousin | 1 in 4 (25%) |
No known family history | Carrier chance |
Whites | 1 in 25 (4%) |
Blacks | 1 in 65 (1.5%) |
Asians | 1 in 150 (0.67%) |
In the pancreas, fibrotic tissue, multiple cysts, thick mucus, and eventually fat replace the acini (small, saclike cluster of cells normally found in this gland), producing symptoms of pancreatic insufficiency: altered pancreatic enzymes and insufficient insulin production, abnormal glucose tolerance, and glycosuria. About 15% of patients have adequate pancreatic exocrine function for normal digestion and, therefore, have a better prognosis. Biliary obstruction and fibrosis may prolong neonatal jaundice. In some patients, cirrhosis and portal hypertension may lead to esophageal varices, episodes of hematemesis and, occasionally, hepatomegaly.
DIAGNOSIS
The Cystic Fibrosis Foundation has developed certain criteria for a definitive diagnosis: two sweat tests using a pilocarpine solution (a sweat inducer) and either obstructive pulmonary disease, confirmed pancreatic insufficiency or failure to thrive, and a family history of cystic fibrosis.
The following test results may support the diagnosis:
Chest X-rays indicate early signs of obstructive lung disease.
Stool specimen analysis indicates the absence of trypsin, suggesting pancreatic insufficiency.
Deoxyribonucleic acid testing can now locate the presence of the deltaF508 deletion (found in about 70% of cystic fibrosis patients, although the disease can cause more than 100 other mutations). It allows prenatal diagnosis in families with a previously affected child.
Pulmonary function tests reveal decreased vital capacity, elevated residual volume due to air entrapments, and decreased forced expiratory volume in 1 second. This test is used if pulmonary exacerbation already exists.
Liver enzyme tests may reveal hepatic insufficiency.
Sputum culture reveals organisms that cystic fibrosis patients typically and chronically colonize, such as Staphylococcus and Pseudomonas.
Serum albumin measurement helps assess nutritional status.
Electrolyte analysis assesses hydration status.
TREATMENT
The aim of treatment is to help the child lead as normal a life as possible. The type of treatment depends on the organ systems involved.
To combat electrolyte losses in sweat, salt foods generously and, in hot weather, administer sodium supplements.
To offset pancreatic enzyme deficiencies, give oral pancreatic enzymes with meals and snacks, as ordered. Maintain a diet that’s low in fat, but high in protein and calories, and provide supplements of water-miscible, fat-soluble vitamins (A, D, E, and K).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


