Overview
There are three major types of mutations: (1) genome mutations, which involve loss or gain of an entire chromosome; (2) chromosomal mutations, which involve alterations in one or more chromosomes that are usually identifiable by karyotyping; and (3) gene mutations, which are partial or complete deletion of the gene or alteration of the base. Genome mutations usually result in death of the fetus, or death during infancy or early childhood.
Many diseases have a genetic component, albeit without a specific identifiable gene mutation. Such conditions are said to have a multifactorial inheritance pattern. Examples of such diseases include coronary artery disease, hypertension, gout, and diabetes mellitus.
When discussing genetic diseases, some definitions are important to remember: (1) hereditary or familial, a condition derived from parents (i.e., a condition that is transmitted in the germ line); and (2) congenital, a condition that is present at birth. Not all hereditary conditions are congenital, and not all congenital conditions are hereditary. Some hereditary conditions are manifested at the time of birth or shortly thereafter, and many manifest later in life.
The overall effects of the mutation of a single gene include (1) an enzyme defect; (2) defects in membrane receptors and/or transport system; (3) alterations in structure, function, or quantity of nonenzymatic protein; or (4) mutations resulting in unusual reactions to drugs. An enzyme defect can cause accumulation of substrate, a metabolic block resulting in a decreased amount of needed end product, or failure to inactivate a tissue-damaging substrate.
Autosomal Dominant Disorders
Overview: In general, autosomal dominant disorders have reduced penetrance and variable expressivity. They usually do not encode enzymes because a loss of up to 50% of an enzyme’s activity can be compensated for by activity of the enzyme encoded by the normal allele (Table 6-1).
Disorder | Protein Involved |
|---|---|
Familial hypercholesterolemia | LDL receptor |
Familial adenomatous polyposis | APC protein |
Hereditary spherocytosis | Ankyrin, spectrin, band 4.1, or band 3 |
Marfan syndrome | Fibrillin-1 |
Neurofibromatosis, type 1 | Neurofibromin |
Neurofibromatosis, type 2 | Merlin |
Tuberous sclerosis | Hamartin, tuberin |
Mutation: Low-density lipoprotein receptor gene (LDL); there are more than 100 known mutations.
Mechanism: The LDL receptor recognizes apolipoprotein B100 or apolipoprotein E; therefore, a mutation of the receptor results in impaired uptake of cholesterol into cells.
Manifestations of familial hypercholesterolemia
- Elevated cholesterol level: Heterozygotes have half the normal amount of LDL receptors and two to three times the normal level of cholesterol; homozygotes have five or more times the normal level of cholesterol.
- Tendon sheath xanthomas, corneal arcus, and xanthelasma.
- Early atherosclerosis and its consequences; homozygotes usually die of cardiovascular disease before the age of 30 years.
Mutation: APC gene on chromosome 5q21.
Mechanism: The APC (adenomatous polyposis coli) protein degrades β-catenin (accumulated β-catenin activates transcription of genes such as MYC and cyclin D1)—therefore, by allowing β-catenin to accumulate, a mutation of the APC gene promotes cell proliferation.
Manifestations of FAP: Multiple (> 100) colonic polyps and congenital hypertrophy of the retinal pigment epithelia. A subset of patients with FAP develops various extraintestinal manifestations such as osteomas and soft tissue tumors (Figure 6-1).
Figure 6-1.
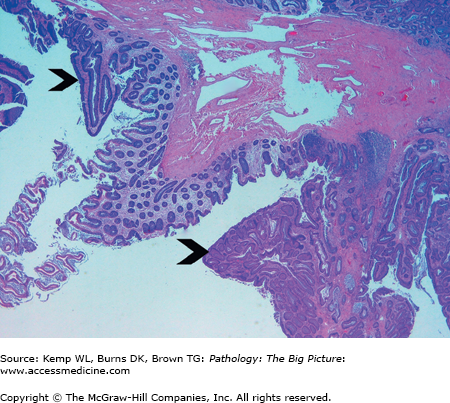
Familial adenomatous polyposis. Patients with this hereditary condition develop hundreds of adenomas in their colon, and eventually develop invasive colonic adenocarcinoma. This low-power view of the colon shows two tubular adenomas (arrowheads) with intervening normal colonic mucosa. Hematoxylin and eosin, 20×.
Complications: Development of invasive colonic adenocarcinoma in all patients by the fifth decade of life.
Inheritance pattern: Can be autosomal dominant or autosomal recessive; the autosomal recessive form is more severe, however.
Mutation: Gene for ankyrin, spectrin, band 4.1, or band 3.
Manifestations of hereditary spherocytosis
- Anemia, due to extravascular hemolysis because affected red blood cells do not deform well in transit through the spleen.
- Splenomegaly due to sequestration of abnormal red blood cells in the spleen.
- Cholelithiasis caused by increased production of bile pigments from hemolysis of red blood cells.
Laboratory testing: Increased osmotic fragility.
Treatment of hereditary spherocytosis: Splenectomy.
Mutation: Fibrillin-1 gene on chromosome 15q21.
Mechanism: Fibrillin provides support for deposition of tropoelastin and production of elastic fibers.
Manifestations of Marfan syndrome by organ system
- Skeletal: Long arms and legs and long fingers (arachnodactyly).
- Eye: Bilateral dislocation of the lens (ectopia lentis).
- Cardiovascular
- Aortic root dilation, leading to aortic insufficiency.
- Myxomatous mitral valve.
- Ascending thoracic aortic aneurysms.
- Aortic dissection associated with cystic medial degeneration.
- Aortic root dilation, leading to aortic insufficiency.
NF-1
Mutation: NF-1 gene on 17q11.2; there is a high rate of spontaneous mutations (occurring in roughly 50% of new cases of NF-1).
Mechanism: The NF-1 gene product, neurofibromin, is a guanosine triphosphatase (GTPase)-activating protein that influences normal Schwann cell proliferation and differentiation. Although its role remains incompletely understood, it is thought to act as a tumor suppressor protein.
Incidence: 1 in 3000 individuals in the general population.
Manifestations of NF-1 (Figure 6-2)
- Neurofibromas (proliferation of Schwann cells, perineurial cells, and fibroblasts): The three types of neurofibromas are cutaneous, subcutaneous, and plexiform.
- Café-au-lait spots (pigmented cutaneous macules).
- Lisch nodules (iris hamartomas).
- Optic gliomas (e.g., pilocytic astrocytomas).
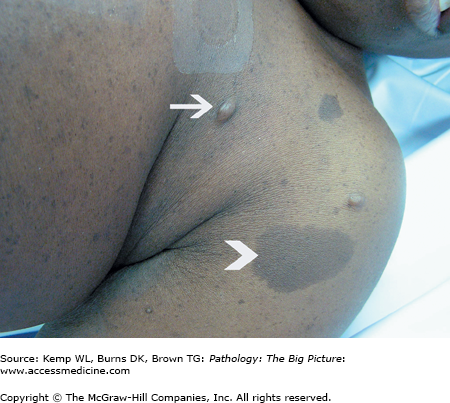
Important points regarding NF-1: Plexiform neurofibromas can transform into malignant peripheral nerve sheath tumors; in contrast, malignant transformation of superficial neurofibromas is extremely rare (Figure 6-3).
Figure 6-3.

Malignant peripheral nerve sheath tumor. This patient had type 1 neurofibromatosis. A plexiform neurofibroma in the pleural cavity underwent malignant degeneration, resulting in a malignant peripheral nerve sheath tumor. The tumor encased the left lung and heart, with only a small focus of invasion into the pulmonary parenchyma (not in the photograph). The photograph shows a bisected left lung with the tumor at its periphery.
NF-2
Mutation: NF-2 gene on chromosome 22q12.
Mechanism: The NF-2 gene product merlin is a protein that binds to cellular membranes and cytoskeletal components (particularly actin) and plays a role in regulating contact inhibition and proliferation of Schwann cells. Like the NF-1 gene, the NF-2 gene is thought to act as a tumor suppressor gene.
Incidence: 1 in 40,000 to 50,000 in the general population.
Manifestations of NF-2: Bilateral acoustic schwannomas, multiple meningiomas.
Mutation: VHL gene on chromosome 3p25.
Associated neoplasms: Renal cell carcinoma, pheochromocytomas, hemangioblastomas of the central nervous system, and retinal angiomas.
Mutation: Gene for hamartin on chromosome 9 or tuberin on chromosome 16.
Manifestations
- Cerebral cortical hamartomas (tubers).
- Subependymal giant cell astrocytomas (Figure 6-4).
- Seizures and mental retardation.
- Cardiac rhabdomyomas.
- Renal angiomyolipomas.
- Localized leathery thickenings of the skin (i.e., shagreen patches).
- Hypopigmented regions of skin (i.e., ash-leaf patches).
- Adenoma sebaceum.
Mutation: Expanded CTG trinucleotide repeats on chromosome 19q13.2-13.3, which affects mRNA for DMPK (dystrophica myotonia-protein kinase); normal individuals have less than 30 repeats. The more repeats, the more severely affected a patient will be.
Manifestations of myotonic dystrophy type 1
- The classic hallmark for clinical presentation is the inability to relax the muscles after stimulation. Patients often describe inability to release a handshake.
- Weakness is common.
- Atrophy of facial muscles (“hatchet-shaped facies”).
- Cataracts, gonadal atrophy, and cardiac problems (especially conduction defects and arrhythmias).
Autosomal Recessive Disorders
Overview: Patients with autosomal recessive disorders usually have complete penetrance and more uniform expression of the disease when compared to patients with autosomal dominant disorders. Autosomal recessive disorders often involve enzymes (Table 6-2).
Disorder | Protein Involved |
|---|---|
Cystic fibrosis | Cystic fibrosis transmembrane conductance regulator |
Galactosemia | Galactose-1-phosphate uridyltransferase |
Hereditary hemochromatosis | HFE protein |
Phenylketonuria | Phenylalanine hydroxylase |
Wilson disease | ATP-dependent Cu2+ transporter |
Alkaptonuria | Homogentisic oxidase |
Maple syrup urine disease | Branched chain ketoacid dehydrogenase |
Hurler disease | α-L-iduronidase |
Hunter disease | Iduronate-2 sulfatase |
Tay-Sachs disease | α-subunit of GM2 gangliosidase |
Gaucher disease | Glucocerebrosidase |
Niemann-Pick, types A and B | Sphingomyelinase |
Genetic abnormality: The gene associated with this condition is found on chromosome 14; the normal allele is PiMM. Patients with PiZZ alleles have circulating levels of AAT that are only 10% of normal.
Manifestations
- Panacinar emphysema, which is dramatically accelerated by cigarette smoking.
- Cirrhosis—only 10% or more of individuals with PiZZ alleles develop cirrhosis, most likely because they have decreased protein degradation via the ubiquitin-proteosome pathway. Liver biopsy shows periodic acid–Schiff (PAS) positive and diastase-resistant granules, which represent accumulations of an abnormal AAT.



