Introduction
Mechanisms of cellular and tissue dysfunction in genetic diseases are as varied as the organs they affect. To some extent, these mechanisms are similar to those that occur in nonheritable disorders. For example, a fracture resulting from decreased bone density in osteoporosis heals in much the same way as one caused by a defective collagen gene in osteogenesis imperfecta, and the response to coronary atherosclerosis in most individuals does not depend on whether they have inherited a defective low-density lipoprotein (LDL) receptor. Thus, the pathophysiologic principles that distinguish genetic disease focus not so much on the affected organ system as on the mechanisms of mutation, inheritance, and molecular pathways from genotype to phenotype.
This chapter begins with a discussion of the terminology used to describe inherited conditions, the prevalence of genetic disease, and some major principles and considerations in medical genetics. Important terms and key words used throughout the chapter are defined in Table 2–1.
| Term | Definition |
|---|---|
| Acrocentric | Refers to the terminal location of the centromere on chromosomes 13, 14, 15, 21, and 22. |
| Allelic heterogeneity | The situation in which multiple alleles at a single locus can produce one or more disease phenotypes. |
| Amorphic | Refers to mutations that cause a complete loss of function for the respective gene, and therefore yield the same phenotype as a complete gene deletion. |
| Aneuploidy | A general term used to denote any unbalanced chromosome complement. |
| Antimorphic | Refers to mutations that when present in heterozygous form opposite a nonmutant allele will result in a phenotype similar to homozygosity for loss-of-function alleles. |
| Ascertainment bias | The situation in which individuals or families in a genetic study are not representative of the general population because of the way in which they are identified. |
| Autosomal | Located on chromosomes 1–22 rather than X or Y. |
| CpG island | A segment of DNA that contains a relatively high density of 5′- CG-3′ dinucleotides. Such segments are frequently unmethylated and located close to ubiquitously expressed genes. |
| Dictyotene | The end of prophase during female meiosis I in which fetal oocytes are arrested prior to ovulation. |
| Dominant | A pattern of inheritance or mechanism of gene action in which the effects of a variant allele can be observed in the presence of a nonmutant allele. |
| Dominant negative | A type of pathophysiologic mechanism that occurs when a mutant allele interferes with the normal function of the nonmutant gene product. |
| Dosage compensation | Mechanism by which a difference in gene dosage between two cells is equalized. For XX cells in mammals, decreased expression from one of the two X chromosomes results in a concentration of gene product similar to an XY cell. |
| End-product deficiency | A pathologic mechanism in which absence or reduction in the product of a particular enzymatic reaction leads to disease. |
| Epigenetic | Refers to a phenotypic effect that is heritable, through somatic cell division and/or across organismal generations, but that does not depend on variation in DNA sequence. Instead, epigenetic inheritance is associated with alterations in chromatin structure such as DNA methylation or histone modification that can be transmitted during cell division. |
| Expressivity | The extent to which a mutant genotype affects phenotype, including the tissues that are affected, and the severity of those effects. |
| Fitness | The effect of a mutant allele on an individual’s ability to produce offspring. |
| Founder effect | One of several possible explanations for an unexpectedly high frequency of a deleterious gene in a population. If the population was founded by a small ancestral group, it may have, by chance, contained a large number of carriers for the deleterious gene. |
| Gamete | The egg or sperm cell that represents a potential reproductive contribution to the next generation. Gametes have undergone meiosis and so contain half the normal number of chromosomes found in zygotic cells. |
| Gene dosage | The principle that the amount of product expressed for a particular gene is proportionate to the number of gene copies present per cell. |
| Genetic anticipation | A clinical phenomenon in which the phenotype observed in individuals carrying a deleterious gene appears more severe in successive generations. Possible explanations include ascertainment bias or a multistep mutational mechanism such as expansion of triplet repeats. |
| Haplotype | A set of closely linked DNA sequence variants on a single chromosome. |
| Hemizygous | A term referring to the presence of only one allele at a locus, either because the other allele is deleted or because it is normally not present, e.g., X-linked genes in males. |
| Heterochromatin | One of two alternative forms of chromosomal material (the other is euchromatin) in which chromosomal DNA is highly condensed and, usually, devoid of genes that are actively transcribed. |
| Heteroplasmy | The mixture of mutant and nonmutant mitochondrial DNA molecules in a single cell. |
| Heterozygote advantage | One way to explain an unexpectedly high frequency of a recessively inherited mutation in a particular population. During recent evolution, carriers (i.e., heterozygotes) are postulated to have had a higher fitness than homozygous nonmutant individuals. |
| Heterozygous | Having two alleles at the same locus that are different. |
| Homozygous | Having two alleles at the same locus that are the same. |
| Hypermorphic | Refers to a mutation that has an effect similar to increasing the number of normal gene copies per cell. |
| Hypomorphic | Refers to a mutation that reduces but does not eliminate the activity of a particular gene product. |
| Imprinting | Most commonly, the process whereby expression of a gene depends on whether it was inherited from the mother or the father. |
| Linkage disequilibrium | A condition in which certain combinations of closely linked alleles, or haplotypes, are present in a population at frequencies not predicted by their individual allele frequencies. |
| Locus heterogeneity | A situation in which mutations of different genes produce similar or identical phenotypes. Also referred to as genetic heterogeneity. |
| Mendelian | A form of inheritance that obeys Mendel laws, ie, autosomal dominant, autosomal recessive, X-linked dominant, or X-linked recessive. |
| Mosaicism | A situation in which a genetic alteration is present in some but not all of the cells of a single individual. In germline or gonadal mosaicism, the alteration is present in germ cells but not somatic cells. In somatic mosaicism, the genetic alteration is present in some but not all of the somatic cells (and is generally not present in the germ cells). |
| Monosomy | A reduction in zygotic cells from two to one in the number of copies for a particular chromosomal segment or chromosome. |
| Neomorphic | Refers to a mutation that imparts a novel function to its gene product and consequently results in a phenotype distinct from an alteration in gene dosage. |
| Nondisjunction | Failure of two homologous chromosomes to separate, or disjoin, at metaphase of meiosis I, or the failure of two sister chromatids to disjoin at metaphase of meiosis II or mitosis. |
| Penetrance | In a single individual of a variant genotype, penetrance refers to whether or not the variant genotype can be inferred based on defined phenotypic criteria. In a population, reduced penetrance refers to the rate at which individuals of a variant genotype cannot be recognized according to specific phenotypic criteria. |
| Phenotypic heterogeneity | The situation that pertains when mutations of a single gene produce multiple different phenotypes. |
| Postzygotic | A mutational event that occurs after fertilization and that commonly gives rise to mosaicism. |
| Premutation | A genetic change that does not result in a phenotype itself but has a high probability of developing a second alteration—a full mutation—which does cause a phenotype. |
| Primordial germ cell | The group of cells set aside early in development that go on to give rise to gametes. |
| Recessive | A pattern of inheritance or mechanism of gene action in which a particular mutant allele causes a phenotype only in the absence of a nonmutant allele. Thus, for autosomal conditions, the variant or disease phenotype is manifest when two copies of the mutant allele are present. For X-linked conditions, the variant or disease phenotype is manifest in cells, tissues, or individuals in which the nonmutant allele is either inactivated (a heterozygous female) or not present (a hemizygous male). |
| Robertsonian translocation | A type of translocation in which two acrocentric chromosomes are fused together with a single functional centromere. A carrier of a robertsonian translocation with 45 chromosomes has a normal amount of chromosomal material and is said to be euploid. |
| SNP | Single nucleotide polymorphism—one of the most common types of genetic variation. There are approximately 1 million common SNPs in the human genome (those that exist at a frequency >1%), and billions of rare single-nucleotide variants (at a frequency >0.001%). Most do not affect protein structure, but the common SNPs may serve as valuable markers for determining the effect of genetic variation on complex and common diseases and disorders such as diabetes, heart disease, hypertension, and obesity. |
| Structural variant | A deletion, insertion, or more complex rearrangement, usually caused by recombination between repetitive elements. Also referred to as copy number variant (CNV) and the most common type of genomic variation. Most structural variants involve deletions or insertions that are relatively small (<10 kb) and do not cause any clinical phenotype. Larger structural variants (>100 kb) are increasingly likely to have clinical effects. |
| Substrate accumulation | A pathogenetic mechanism in which deficiency of a particular enzyme causes disease because the substrate of that enzyme accumulates in tissue or blood. |
| Triplet repeat | A three-nucleotide sequence that is tandemly repeated many times—ie, (XYZ)n. Alterations in length of such simple types of repeats (dinucleotide and tetranucleotide as well) occur much more frequently than most other kinds of mutations; in addition, alteration in the length of trinucleotide repeats is the molecular basis for several heritable disorders. |
| Trisomy | An abnormal situation in which there are three instead of two copies of a chromosomal segment or chromosome per cell. |
Next, a group of disorders caused by mutations in collagen genes is discussed (ie, osteogenesis imperfecta). Although osteogenesis imperfecta is often considered a single entity, different mutations and different genes subject to mutation lead to a wide spectrum of clinical phenotypes. The different types of osteogenesis imperfecta exhibit typical patterns of autosomal dominant or autosomal recessive inheritance and are, therefore, examples of so-called mendelian conditions. To show how environmental factors can influence the relationship between genotype and phenotype, I discuss another mendelian condition, phenylketonuria. This serves as a paradigm for newborn screening programs and treatment of genetic disease. Several genetic conditions have been found to depend not only on the gene being inherited but also on the phenotype or the sex of the parent. As an example of a condition that exhibits nontraditional inheritance, fragile X–associated mental retardation syndrome is discussed. This syndrome not only is the most common inherited cause of mental retardation but also illustrates how different types of mutations can explain the perplexing phenomenon of genetic anticipation, where the severity of a mendelian syndrome appears to progress with every generation of inheritance. Another group of disorders that depend on the phenotype and sex of the parent consists of those that affect the mitochondrial genome. As examples, Leber hereditary optic neuropathy (LHON) and myoclonic epilepsy with ragged red fibers (MERRF) are considered. These illustrate the principles of mitochondrial inheritance and its pathophysiology. Aneuploidy is discussed as one of the most common types of human genetic disease that does not affect DNA structure but instead alters the normal chromosome content per cell. The example that is considered, Down syndrome, has had a major impact on reproductive medicine and reproductive decision making and serves to illustrate general principles that apply to many aneuploid conditions. Finally, I consider how genome sequences and sequencing are improving our understanding of pathophysiology for many diseases. With the completion of the human genome sequence and technological advances that allow individual genomes to be sequenced rapidly and inexpensively, prospects are at hand to identify genetic components of any human phenotype and to provide medical care that is truly personalized.
Unique Pathophysiologic Aspects of Genetic Diseases
Although the phenotypes of genetic diseases are diverse, their causes are not. The primary cause of any genetic disease is a change in the sequence or cellular content of DNA that ultimately deranges gene expression. Most genetic diseases are caused by an alteration in DNA sequence that alters the synthesis of a single gene product. However, some genetic diseases are caused by (1) structural rearrangements that result in deletion or duplication of a group of closely linked genes or (2) abnormalities during mitosis or meiosis that result in an abnormal number of chromosomes per cell. In most genetic diseases, every cell in an affected individual carries the mutated gene or genes as a consequence of its inheritance via a mutant egg or sperm cell (gamete). However, mutation of the gametic cell may have arisen during its development, in which case somatic cells of the parent do not carry the mutation and the affected individual is said to have a “new mutation.” In addition, some mutations may arise during early embryogenesis, in which case tissues of the affected individual contain a mixture, or mosaic, of mutant and nonmutant cells. Depending on the time of embryogenesis and cell type in which a new mutation arises, an individual may carry the mutation in some but not all of their germ cells (germline mosaicism), some but not all of their somatic cells (somatic mosaicism), or both.
It is helpful to begin with a brief review of terms that are commonly used in discussing genetic disease with patients and their families. Although genes were recognized and studied long before the structure of DNA was known, it has become common usage to regard a gene as a short stretch of DNA, usually but not always <100,000 base pairs (bp) in length, that encodes a product (usually protein) responsible for a measurable trait. DNA length is typically measured in base pairs, kilobase pairs (kb), or megabase pairs (Mb); chromosomes vary in length from about 46 Mb to 245 Mb. The locus is the place where a particular gene lies on its chromosome. A gene’s DNA sequence nearly always shows slight differences when many unrelated individuals are compared, and the variant sequences are described as alleles. A mutation is a biochemical event such as a nucleotide change, deletion, or insertion that has produced a new allele. Many changes in the DNA sequence of a gene, such as those within introns or at the third “wobble” position of codons for particular amino acids, do not affect the structure or expression of the gene product; therefore, although all mutations result in a biochemical or molecular biologic phenotype (ie, a change in DNA), only some of them result in a clinically abnormal phenotype.
At the molecular level, variant alleles are usually recognized by DNA sequencing and are referred to as a single nucleotide polymorphism (SNP) if a single base pair change has occurred. As originally coined, the word polymorphism referred to an allele present in 1% or more of a population; today, the terminology tends to be less rigid and is often described qualitatively, ie, rare and common variants. At the clinical level, variant alleles are recognized by their effect on a phenotype such as human leukocyte antigen (HLA) type or hair color. For an autosomal gene (those that lie on chromosomes 1–22, carried in two copies per cell), individuals carrying identical copies are homozygous, whereas individuals whose two copies differ from each other are heterozygous. These terms—homozygous and heterozygous—can apply to the DNA sequence, the protein product, or the clinical phenotype. In other words, an individual may be heterozygous for a SNP that does not alter the protein product, heterozygous for a deletion that causes a genetic disease, or heterozygous for a DNA sequence alteration that causes a change in protein structure but does not cause disease.
This discussion helps to illustrate the use of the word phenotype, which refers simply to any characteristic that can be measured, with the type of measurement depending on the characteristic. Hair color and height are phenotypes readily apparent to a casual observer that are not obviously associated with disease, diabetes and coronary artery disease are disease phenotypes that typically require clinical investigation to be recognized, whereas restriction fragment length polymorphisms (RFLPs), simple sequence length polymorphisms (SSLPs), and SNPs are molecular biologic phenotypes that can be detected only with a laboratory test.
Penetrance & Expressivity
One of the most important principles of human genetics is that two individuals with the same mutated gene may have different phenotypes. For example, in the autosomal dominant condition called type I osteogenesis imperfecta, pedigrees may occur in which there is both an affected grandparent and an affected grandchild even though the obligate carrier parent is asymptomatic (Figure 2–1). Given a set of defined criteria, recognition of the condition in individuals known to carry the mutated gene is described as penetrance. In other words, if 7 of 10 individuals older than 40 with the type I osteogenesis imperfecta mutation have an abnormal bone density scan, the condition is said to be 70% penetrant by that criterion. Penetrance may vary both with age and according to the set of criteria being used; for example, type I osteogenesis imperfecta may be 90% penetrant at age 40 when the conclusion is based on a bone density scan in conjunction with laboratory tests for abnormal collagen synthesis. Reduced penetrance or age-dependent penetrance is a common feature of dominantly inherited conditions that have a relatively high fitness (the extent to which individuals carrying a mutant allele produce offspring relative to individuals who do not carry a mutant allele); Huntington disease and polycystic kidney disease are examples.
Figure 2–1
Penetrance and expressivity in type I osteogenesis imperfecta. In this schematic pedigree of the autosomal dominant condition type I osteogenesis imperfecta, nearly all of the affected individuals exhibit different phenotypic features that vary in severity (variable expressivity). As is shown, type I osteogenesis imperfecta is fully penetrant, because every individual who transmits the mutation is phenotypically affected to some degree. However, if mild short stature in the individual indicated with the arrow had been considered to be a normal variant, then the condition would have been nonpenetrant in this individual. Thus, in this example, judgments about penetrance or nonpenetrance depend on the criteria for normal and abnormal stature.
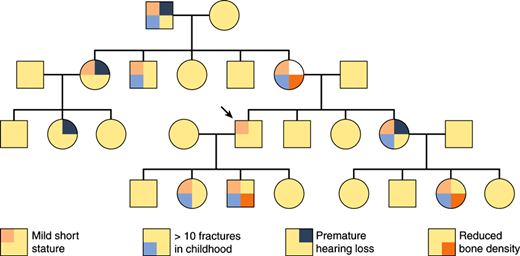
When the same mutated gene gives rise to a different spectrum of phenotypes, the situation is referred to as variable expressivity. For example, blue scleras and short stature may be the only manifestations of type I osteogenesis imperfecta in a particular individual, whereas a sibling who carries the identical mutation may be confined to a wheelchair as a result of multiple fractures and deformities. The mutation is penetrant in both individuals, but its expression is variable. Both reduced penetrance and variable expressivity may occur in individuals who carry the same mutated allele; therefore, phenotypic differences between these individuals must be due to the effects of other “modifier” genes, to environmental interactions, or to chance.
Mechanisms of Mutation & inheritance patterns
Mutations can be characterized both by their molecular nature—nucleotide deletion, insertion, substitution—and by their effects on gene activity (ie, no effect [neutral or silent], complete loss of function [amorphic mutation], partial loss of function [hypomorphic mutation], gain of function [hypermorphic mutation], or acquisition of a new property [neomorphic mutation]). Geneticists who study experimental organisms frequently use specific deletions to ensure that a mutated allele causes a loss of function, but human geneticists rely on biochemical or cell culture studies. Amorphic and hypomorphic mutations are probably the most frequent type of mutation in human genetic disease because there are many ways to interfere with a protein’s function.
For autosomal genes, the fundamental difference between dominant and recessive inheritance is that, with dominant inheritance, the disease state or trait being measured is apparent when one copy of the mutated allele and one copy of the normal allele are present. With recessive inheritance, two copies of the mutated allele must be present for the disease state or trait to be apparent. However, for genes that lie on the X chromosome, the situation is slightly different because females have two X chromosomes and males have only one. X-linked dominant inheritance occurs when one copy of a mutant gene causes the disease phenotype (in males and females); X-linked recessive inheritance occurs when two copies of a mutant gene cause the disease phenotype (in females). Because most mutations are amorphic or hypomorphic, however, one copy of an X-linked mutant allele in males is not “balanced” with a nonmutant allele, as it would be in females; therefore, in X-linked recessive inheritance, one copy of a mutant allele is sufficient to produce a disease phenotype in males, a situation referred to as hemizygosity.
Most recessive mutations are due to loss of function of the gene product, which can occur from a variety of different causes, including failure of the gene to be transcribed or translated and failure of the translated gene product to function correctly. There are two general principles to keep in mind when considering loss-of-function mutations. First, because expression from the nonmutant allele usually does not change (i.e., there is no dosage compensation), gene expression in a heterozygous carrier of a loss-of-function allele is reduced to 50% of normal. Second, for most biochemical pathways, a 50% reduction in enzyme concentration is not sufficient to produce a disease state. Thus, most diseases resulting from enzyme deficiencies such as phenylketonuria (Table 2–2) are inherited in a recessive fashion.
| Disorder | Phenotype | Genetic Mechanism | Incidence |
|---|---|---|---|
| Down syndrome | Mental and growth retardation, dysmorphic features, internal organ anomalies | Chromosomal imbalance; caused by trisomy 21 | ≈1:800; increased risk with advanced maternal age |
| Fragile X–associated mental retardation | Mental retardation, characteristic facial features, large testes | X-linked; progressive expansion of unstable DNA causes failure to express gene encoding RNA-binding protein | ≈1:1500 males; can be manifested in females; multistep mechanism |
| Sickle cell anemia | Recurrent painful crises, increased susceptibility to infections | Autosomal recessive; caused by a single missense mutation in beta-globin | ≈1:400 blacks |
| Cystic fibrosis | Recurrent pulmonary infections, exocrine pancreatic insufficiency, infertility | Autosomal recessive; caused by a multiple loss-of-function mutations in a chloride channel | ≈1:2000 whites; very rare in Asians |
| Leber hereditary optic neuropathy | Acute or subacute blindness, occasional myopathy or neurodegeneration | Mutation of electron transport chain encoded by mtDNA | ≈1:50,000–1:10,000 |
| Myoclonic epilepsy with ragged red fibers | Uncontrolled periodic jerking, muscle weakness | Mutation of mitochondrial tRNA in mtDNA | ≈1:100,000–1:50,000 |
| Neurofibromatosis | Multiple café-au-lait spots, neurofibromas, increased tumor susceptibility | Autosomal dominant; caused by multiple loss-of-function mutations in a signaling molecule | ≈1:3000; ≈50% are new mutations |
| Duchenne muscular dystrophy | Muscular weakness and degeneration | X-linked recessive; caused by multiple loss-of-function mutations in muscle protein | ≈1:3000 males; ≈33% are new mutations |
| Osteogenesis imperfecta | Increased susceptibility to fractures, connective tissue fragility, blue scleras | Phenotypically and genetically heterogeneous | ≈1:10,000 |
| Phenylketonuria | Mental and growth retardation | Autosomal recessive; caused by multiple loss-of-function mutations in phenylalanine hydroxylase | ≈1:10,000 |
If 50% of a particular product is not enough for the cell or tissue to function normally, then a loss-of-function mutation in this gene produces a dominantly inherited phenotype. Such mutations often occur in structural proteins; an example is type I osteogenesis imperfecta, which is considered in detail later. Most dominantly inherited phenotypes are actually semidominant, which means that an individual who carries two copies of the mutant allele is affected more severely than someone who carries one mutant and one normal copy. However, for most dominantly inherited conditions, homozygous mutant individuals are rarely observed. For example, inheritance of achondroplasia, the most common genetic cause of very short stature, is usually described as autosomal dominant. However, rare matings between two affected individuals have a 25% probability of producing offspring with two copies of the mutant gene. This results in homozygous achondroplasia, a condition that is very severe and usually fatal in the perinatal period; thus, achondroplasia exhibits semidominant inheritance. Huntington disease, a dominantly inherited neurologic disease, is the only known human condition in which the homozygous mutant phenotype is identical to the heterozygous mutant phenotype (sometimes referred to as a “true dominant”).
A special kind of pathophysiologic mechanism, referred to as dominant negative, occurs frequently in human genetic diseases that involve proteins that form oligomeric or polymeric complexes. In these disorders, the mutant allele gives rise to a structurally abnormal protein that interferes with the function of the normal allele. Note that any molecular lesion (ie, deletion, nonsense, missense, or splicing) can produce a loss-of-function allele. However, only molecular lesions that yield a protein product (ie, splicing, missense, or nonsense mutations) can result in a dominant negative allele. Type II osteogenesis imperfecta, described later, is an example of a dominant negative mutation.
Although the terms “dominant” and “recessive” are occasionally used to describe specific mutations, a DNA sequence alteration itself cannot, strictly speaking, be dominant or recessive. The terms are instead appropriate to the effect of a mutation on a particular trait. Therefore, in characterizing a particular mutation as “recessive,” one is referring to the effect of the mutation on the trait being studied.
Mutation Rate & The Prevalence of Genetic Disease
At the level of DNA sequence, nucleotide mutations (substitutions, small insertions, or small deletions) in humans occur at a rate of approximately 2 × 10−8 per nucleotide per human generation, or 150 new mutations per diploid genome. However, only about 5% of the human genome is functional, so most new mutations have no effect. Still, with approximately 23,000 genes in the human genome and an estimated deleterious “per locus” mutation rate of 10−5 per generation, the chance of a new deleterious mutation in any one individual is about 20%. Furthermore, assuming 10 billion new births in the last millennium, every gene in the human genome has probably been mutated (in a deleterious manner) about 100,000 different times. However, from a clinical perspective, only about 5000 single-gene disorders have been recognized to cause a human disease. In considering possible explanations for this disparity, it seems likely that deleterious mutations of many single genes are lethal very early in development and thus not clinically apparent, whereas deleterious mutations in other genes do not cause an easily recognizable phenotype. The overall frequency of disease attributable to defects in single genes (ie, mendelian disorders) is approximately 1% of the general population.
Table 2–2 lists the major symptoms, genetic mechanisms, and prevalence of the diseases considered in this chapter as well as of several others. The most common conditions, such as neurofibromatosis, cystic fibrosis, and fragile X–associated mental retardation syndrome, will be encountered at some time by most health care professionals regardless of their field of interest. Other conditions such as Huntington disease and adenosine deaminase deficiency, although of intellectual and pathophysiologic interest, are not likely to be seen by most practitioners.
Many common conditions such as atherosclerosis and breast cancer that do not show strictly mendelian inheritance patterns have a genetic component evident from familial aggregation or twin studies. These conditions are usually described as multifactorial, which means that the effects of one or more mutated genes and environmental differences all contribute to the likelihood that a given individual will manifest the phenotype.
Issues in Clinical Genetics
Most patients with genetic disease present during early childhood with symptoms that ultimately give rise to a diagnosis such as fragile X–associated mental retardation or Down syndrome. The major clinical issues at presentation are arriving at the correct diagnosis and counseling the patient and family regarding the natural history and prognosis of the condition. It is important to assess the likelihood that the same condition will occur again in the family and determine whether it can be diagnosed prenatally. These issues are the subject matter of genetic counseling by medical geneticists and genetic counselors.
Understanding the pathophysiology of genetic diseases that interfere with specific metabolic pathways—so-called inborn errors of metabolism—has led to effective treatments for selected conditions such as phenylketonuria, maple syrup urine disease, and homocystinuria. Many of these diseases are rare, but efforts are underway to develop treatments for common single-gene disorders such as Duchenne muscular dystrophy, cystic fibrosis, and hemophilia. Some forms of therapy are directed at replacing the mutant protein, whereas others are directed at ameliorating its effects.
Checkpoint
Define gene, locus, allele, mutation, heterozygosity, hemizygosity, polymorphism, and phenotype.
How is it possible for two individuals with the same mutation to have differences in the severity of an abnormal phenotype?
Explain the pathophysiologic difference between mutations that act via loss of function and those that act via dominant negative gene action.
Pathophysiology of Selected Genetic Diseases
Osteogenesis imperfecta is a condition inherited in mendelian fashion that illustrates many principles of human genetics. It is a heterogeneous and pleiotropic group of disorders characterized by a tendency toward fragility of bone. Advances in the last two decades demonstrate two genetically different groups: the “classical” group, in which more than 90% of cases are caused by a mutation of the COL1A1 or COL1A2 genes, which encode the subunits of type I collagen, proα1(I) and proα2(I), respectively, and a newer group, caused by loss-of-function mutations in proteins required for proper folding, processing, and secretion of collagen. More than 100 different mutant alleles have been described for osteogenesis imperfecta; the relationships between different DNA sequence alterations and the type of disease (genotype-phenotype correlations) illustrate several pathophysiologic principles in human genetics.
The clinical and genetic characteristics of osteogenesis imperfecta are summarized in Table 2–3, in which the timing and severity of fractures, radiologic findings, and presence of additional clinical features help to distinguish four different subtypes. This classification was presented more than 30 years ago. Over the past decade, it has become clear that there are more than a dozen different genes in which mutations can cause osteogenesis imperfecta, and that the utility of alternative or more extended nosologic approaches depends on whether the condition is being considered from the perspective of patients, caregivers, or molecular geneticists.
| Type | Phenotype | Genetics | Molecular Pathophysiology |
|---|---|---|---|
| Type I | Mild: Short stature, postnatal fractures, little or no deformity, blue scleras, premature hearing loss | Autosomal dominant | Loss-of-function mutation in proα1(I) chain resulting in decreased amount of mRNA; quality of collagen is normal; quantity is reduced twofold |
| Type II | Perinatal lethal: Severe prenatal fractures, abnormal bone formation, severe deformities, blue scleras, connective tissue fragility | Sporadic (autosomal dominant) | Structural mutation in proα1(I) or proα2(I) chain that has mild effect on heterotrimer assembly; quality of collagen is severely abnormal; quantity often reduced also |
| Type III | Progressive deforming: Prenatal fractures, deformities usually present at birth, very short stature, usually nonambulatory, blue scleras, hearing loss | Autosomal dominant 1 | Structural mutation in proα1(I) or proα2(I) chain that has mild effect on heterotrimer assembly; quality of collagen is severely abnormal; quantity can be normal |
| Type IV | Deforming with normal scleras: Postnatal fractures, mild-to-moderate deformities, premature hearing loss, normal or gray scleras, dental abnormalities imperfect | Autosomal dominant | Structural mutation in the proα2(I), or, less frequently, proα1(I) chain that has little or no effect on heterotrimer assembly; quality of collagen is usually abnormal; quantity can be normal |
All forms of osteogenesis imperfecta are characterized by increased susceptibility to fractures (“brittle bones”), but there is considerable phenotypic heterogeneity, even within individual subtypes. Individuals with type I or type IV osteogenesis imperfecta present in early childhood with one or a few fractures of long bones in response to minimal or no trauma; x-ray films reveal mild osteopenia, little or no bony deformity, and often evidence of earlier subclinical fractures. However, most individuals with type I or type IV osteogenesis imperfecta do not have fractures in utero. Type I and type IV osteogenesis imperfecta are distinguished by the severity (less in type I than in type IV) and by scleral hue, which indicates the thickness of this tissue and the deposition of type I collagen. Individuals with type I osteogenesis imperfecta have blue scleras, whereas the scleras of those with type IV are normal or slightly gray. In type I, the typical number of fractures during childhood is 10–20; fracture incidence decreases after puberty, and the main features in adult life are mild short stature, a tendency toward conductive hearing loss, and occasionally dentinogenesis imperfecta. Individuals with type IV osteogenesis imperfecta generally experience more fractures than those with type I and have significant short stature caused by a combination of long bone and spinal deformities, but they often are able to walk independently. Approximately one fourth of the cases of type I or type IV osteogenesis imperfecta represent new mutations; in the remainder, the history and examination of other family members reveal findings consistent with autosomal dominant inheritance.
Type II osteogenesis imperfecta presents at or before birth (diagnosed by prenatal imaging) with multiple fractures, bony deformities, increased fragility of nonbony connective tissue, and blue scleras and usually results in death in infancy. Two typical radiologic findings are the presence of isolated “islands” of mineralization in the skull (wormian bones) and a beaded appearance to the ribs. Nearly all cases of type II osteogenesis imperfecta represent a new dominant mutation, and there is no family history. Death usually results from respiratory difficulties.
Type III osteogenesis imperfecta presents at birth or in infancy with progressive bony deformities, multiple fractures, and blue scleras. It is intermediate in severity between types II and IV; most affected individuals will require multiple corrective surgeries and lose the ability to ambulate by early adulthood. Unlike other forms of osteogenesis imperfecta, which are nearly always due to mutations that act dominantly, type III may be inherited, very rarely, in a recessive manner.
Although different subtypes of osteogenesis imperfecta can often be distinguished biochemically, the classification presented in Table 2–3 is primarily clinical rather than molecular, and the disease phenotypes for each subtype show a spectrum of severities that overlap one another. For example, a few individuals diagnosed with type II osteogenesis imperfecta based on the presence of severe bony deformities in utero will survive for many years and thus overlap the type III subtype. Similarly, some individuals with type IV osteogenesis imperfecta have fractures in utero and develop deformities that lead to loss of ambulation. Distinguishing this presentation from type III osteogenesis imperfecta may be possible only if other affected family members exhibit a milder course.
Additional subtypes of osteogenesis imperfecta have been suggested for individuals that do not match types I–IV, and there are additional disorders associated with congenital fractures that are usually not considered to be “classic” osteogenesis imperfecta. In particular, work over the past several years has identified 10 additional genes in which mutations can cause autosomal recessive osteogenesis imperfecta and has provided additional insight into the genetic pathophysiology. In general, recessively inherited osteogenesis imperfect is caused by loss-of-function mutations in genes whose protein product is required for proper protein folding, intracellular processing, and trafficking of type I collagen.
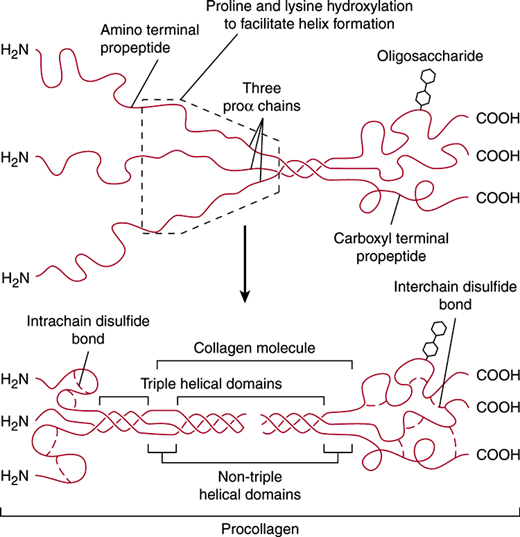
Osteogenesis imperfecta is a disease of type I collagen, which constitutes the major extracellular protein in the body. It is the major collagen in the dermis, the connective tissue capsules of most organs, and the vascular and gastrointestinal (GI) adventitia and is the only collagen in bone. A mature type I collagen fibril is a rigid structure that contains multiple type I collagen molecules packed in a staggered array and stabilized by intermolecular covalent cross-links. Each mature type I collagen molecule contains two α1 chains and one α2 chain, encoded by the COL1A1 and COL1A2 genes, respectively (Figure 2–2). The α1 and α2 chains are synthesized as larger precursors with amino and carboxyl terminal “propeptide” extensions, assemble with each other inside the cell, and are ultimately secreted as a heterotrimeric type I procollagen molecule. During intracellular assembly, the three chains wind around each other in a triple helix that is stabilized by interchain interactions between hydroxylated proline and adjacent carbonyl residues. There is a dynamic relationship between the posttranslational action of prolyl hydroxylase and assembly of the triple helix, which begins at the carboxyl terminal end of the molecule. Increased levels of hydroxylation result in a more stable helix, but helix formation prevents further prolyl hydroxylation. The nature of the triple helix causes the side chain of every third amino acid to point inward, and steric constraints allow only a proton in this position. Thus, the amino acid sequence of virtually all collagen chains in the triple-helical portion is (Gly-X-Y)n, where Y is proline about one third of the time.
Figure 2–2
Molecular assembly of type I pro-collagen. Type I procollagen is assembled in the endoplasmic reticulum from three proα chains that associate with each other beginning at their carboxyl terminals. An important requirement for proper assembly of the triple helix is the presence of a glycine residue at every third position in each of the proα chains. After secretion, the amino and carboxyl terminal propeptides are proteolytically cleaved, leaving a rigid triple helical collagen molecule with very short non–triple-helical domains at both ends. (Modified and reproduced, with permission, from Alberts BA. Molecular Biology of the Cell, 3rd ed. Garland Science, 1994.)
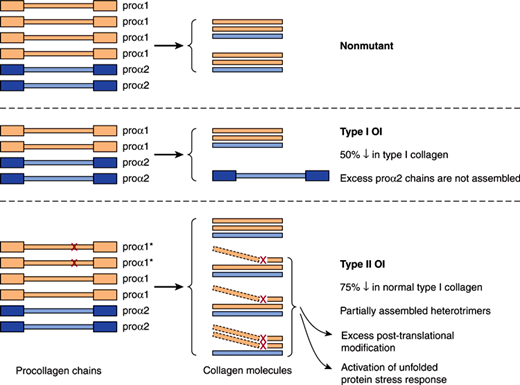
The fundamental defect in most individuals with type I osteogenesis imperfecta is reduced synthesis of type I collagen resulting from loss-of-function mutations in COL1A1. In most cases, the mutant COL1A1 allele gives rise to greatly reduced (partial loss-of-function) or no (complete loss-of-function) mRNA. Because the nonmutant COL1A1 allele continues to produce mRNA at a normal rate (ie, there is no dosage compensation), heterozygosity for a complete loss-of-function mutation results in a 50% reduction in the total rate of proα1(I) mRNA synthesis, whereas heterozygosity for a partial loss-of-function mutation results in a less severe reduction. A reduced concentration of proα1(I) chains limits the production of type I procollagen, leading to (1) a reduced amount of structurally normal type I collagen and (2) an excess of unassembled proα2(I) chains, which are degraded inside the cell (Figure 2–3).
Figure 2–3
Molecular pathogenesis of type I and type II osteogenesis imperfecta (OI). The COL1A1 gene normally produces twice as many proα chains as the COL1A2 gene. Therefore, in nonmutant cells, the ratio of proα1 to proα2 chains is 2:1, which corresponds to the ratio of α1 and α2 chains in intact collagen molecules. In type I osteogenesis imperfecta, a mutation (X) in one of the COL1A1 alleles (*) results in failure to produce proα1 chains, leading to a 50% reduction in the total number of proα1 chains, a 50% reduction in the production of intact type I collagen molecules, and an excess of unassembled proα2 chains, which are degraded inside the cell. In type II osteogenesis imperfecta, a mutation in one of the COL1A1 alleles results in a structural alteration that blocks triple-helix formation and secretion of partially assembled collagen molecules containing the mutant chain. (Adapted from Thompson MW et al. Genetics in Medicine, 5th ed. Saunders, 1991.)
There are several potential molecular defects responsible for COL1A1 mutations in type I osteogenesis imperfecta, including alterations in a regulatory region leading to reduced transcription, splicing abnormalities leading to reduced steady state levels of RNA, and deletion of the entire COL1A1 gene. However, in many cases, the underlying defect is a single base pair change that creates a premature stop codon (also known as a “nonsense mutation”) in an internal exon. In a process referred to as nonsense-mediated decay, partially synthesized mRNA precursors that carry the nonsense codon are recognized and degraded by the cell. With collagen and many other genes, production of a truncated protein (as might be predicted from a nonsense mutation) would be more damaging to the cell than production of no protein at all. Thus, nonsense-mediated decay, which has been observed to occur for mutations in many different multiexon genes, serves as a protective phenomenon and is an important component of the genetic pathophysiology.
An example of these principles is apparent from considering type II osteogenesis imperfecta, which is caused by structurally abnormal forms of type I collagen and is more severe than type I osteogenesis imperfecta. Mutations in type II osteogenesis imperfecta can be caused by defects in either COL1A1 or COL1A2 and usually are missense alterations of a glycine residue that allow the mutant peptide chain to bind to normal chains in the initial steps of trimer assembly (Figure 2–3). However, triple-helix formation is ineffective, often because amino acids with large side chains are substituted for glycine. Ineffective triple-helix formation leads to increased post-translational modification by prolyl hydroxylase, a reduced rate of secretion, and activation of the unfolded protein stress response. These appear to be critical events in the cellular pathogenesis of type II osteogenesis imperfecta, because glycine substitutions toward the carboxyl terminal end of the molecule are generally more severe than those at the amino terminal end.
These considerations help to explain why type II osteogenesis imperfecta is more severe than type I and exemplify the principle of dominant negative gene action. The effects of an amino acid substitution in a proα1(I) peptide chain are amplified at the levels of both triple-helix assembly and fibril formation. Because every type I procollagen molecule has two proα1(I) chains, only 25% of type I procollagen molecules will contain two normal proα1(I) chains even though only one of the two COL1A1 alleles is mutated. Furthermore, activation of the unfolded protein stress response appears to be a key event in the pathophysiology of the disease, as discussed further below. Finally, because each molecule in a fibril interacts with several others, incorporation of an abnormal molecule can have disproportionately large effects on fibril structure and integrity.
Collagen mutations that cause type III and type IV osteogenesis imperfecta are diverse and include glycine substitutions in the amino terminal portion of the collagen triple helix, a few internal deletions of COL1A1 and COL1A2 that do not significantly disturb triple helix formation, and some unusual alterations in the non–triple-helical extensions at the amino and carboxyl terminals of proα chains.
Recessively inherited osteogenesis imperfect can be caused by loss of function for a key prolyl hydroxylase encoded by the PLOD2 gene, one of three genes, CRTAP, LEPRE1, PPIB, that encode members of a protein complex that resides within the rough endoplasmic reticulum and facilitates the folding and processing of type I collagen, as well as several additional genes whose protein products are required for intracellular trafficking and secretion of type I collagen. A common pathway for all types of osteogenesis imperfect involves a combination of reduced production of type I collagen in the extracellular matrix and/or dysfunctional intracellular collagen processing and maturation.
As already described, most cases of type I osteogenesis imperfecta are caused by partial or complete loss-of-function mutations in COL1A1. However, in approximately one-third of affected individuals, the disease is caused by a new mutation; in addition, there are many ways in which DNA sequence alterations can reduce gene expression. Consequently, there is a wide range of mutant alleles (ie, allelic heterogeneity), which represents a challenge for the development of molecular diagnostic tests. In a family in which type I osteogenesis imperfecta is known to occur clinically and a proband seeks a diagnostic test for the purposes of reproductive planning, it is possible in most cases to use linkage analysis at the COL1A1 locus. In this approach, one distinguishes between chromosomes that carry the mutant and nonmutant COL1A1 alleles using closely linked DNA-based polymorphisms, even though the causative molecular defect is not known. Once this information is established for a particular family, inheritance of the mutant allele can be predicted in future pregnancies.
For types III and IV osteogenesis imperfecta, mutations can occur in COL1A1 or COL1A2 (ie, locus heterogeneity), and in this situation, linkage analysis is more difficult because one cannot be sure which locus is abnormal.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


