Chapter 3 A drug is any substance that is used in the diagnosis, cure, treatment, or prevention of a disease or condition (McKenry et al, 2005). This is a general definition that is not very useful because of the great diversity of characteristics and actions of drugs. To be more precise, pharmacokinetics is the study of the action of drugs in the body, including the processes of absorption, distribution, metabolism, and elimination. It may be thought of as what the body does to the drug. Pharmacodynamics is the study of the biochemical and physiologic effects of drugs on the function of living organisms and of their component parts (Brunton, Chabner, & Knollman, 2010). It includes consideration of the mechanisms of drug action and may be viewed as what the drug does to the body. Drugs have three names: a chemical name, a generic name, and a trade, or brand, name. The chemical name describes the drug’s chemical composition and molecular structure. The generic name, or nonproprietary name, is the official name assigned by the manufacturer with the approval of the U.S. Adopted Name Council (USAN) and is the name listed in pharmacology reference books (McKenry et al, 2006). The trade name is the patent name given to the medicine by the company that is marketing the drug. If more than one company manufactures the drug, then the drug will have more than one trade name, thus adding to the confusion. Trade names are usually simpler and easier to remember than generic names because drug companies want you to remember their name and to use their drug. The generic name often will identify the type of drug that it is and thus will reveal the therapeutic effect. For example, it is important to identify a β-blocker by the similar-sounding generic names, such as atenolol, metoprolol, or propranolol, vs. learning the trade names (Tenormin, Lopressor, and Inderal, respectively). Providers should refer to drugs by their generic names when communicating with patients and other health care providers. This is very important with OTC medications because the contents of trade name products may vary. Pharmacokinetics focuses on the processes concerned with absorption, distribution, biotransformation (metabolism), and excretion (elimination) of drugs (Figure 3-1). Absorption describes how the drug leaves its site of administration. The bioavailability of the product is what is most important clinically. Bioavailability is how much of the drug that is administered reaches its site of action (Brunton et al, 2010). The fraction of the drug that reaches the systemic circulation is called the f value. After a solid or liquid drug has been orally ingested, the drug must break up (disintegrate) and then become soluble in body fluids (dissolution) before the process of pharmacokinetic absorption begins (Stringer, 2005). Bioequivalence is an issue with generic vs. trade drugs. Bioequivalence means that two drug products (1) contain the same active ingredients; (2) are identical in strength or concentration, dosage form, and route of administration; and (3) have essentially the same rate and extent of bioavailability (Brunton et al, 2010). Although a generic drug may have the same amount of drug, because of variability in the way the medication is manufactured, slightly more or less drug may be bioavailable. Legally, the drug must be ±20% of the proprietary drug. This variance becomes a concern with certain drugs, especially when the therapeutic window is narrow (e.g., Lanoxin vs. digoxin). Avoiding variation and complying with preferred drug formularies are the major reasons for ordering “Dispense as written” trade name prescriptions. • Formulation of the drug—influences dissolution rate of solid form of drug • Concentration of the drug—the higher the concentration, the more quickly the drug is absorbed • Lipophilic drug formulations are more readily absorbable—Nonionized drugs are more lipid soluble and may readily diffuse across cell membranes. Ionized drugs are lipid insoluble and nondiffusible. • Acidic drugs become nonionized in the acidity of the stomach and then diffuse across membranes—Basic drugs (alkaloids) tend to ionize and are not well absorbed in the stomach, but they may be better absorbed in the small intestine. A change in the pH of the stomach will affect the absorption of many drugs (Katzung, Masters, & Trevor, 2011). • Poor gastrointestinal absorption may occur because of the physical characteristics of the drug. • Irritation to the gastrointestinal mucosa may result in ulceration or emesis. • Destruction of drugs may occur because of digestive enzymes and low gastric pH. • Interactions may occur between the drug and food or other substances in the gastrointestinal tract. Many variations are available in oral drug formulations; in order from the fastest absorption rate to the slowest absorption rate, they include liquids, elixirs, syrups, suspensions, solutions, powders, capsules, tablets, coated tablets, enteric-coated tablets, and slow-release formulations (Brunton et al, 2010; Katzung et al, 2011). Controlled-release preparations are designed to provide slow, uniform absorption of a drug (usually with a short half-life) over a long time, usually from 8 to 12 hours. These work with varying degrees of success. Some formulations are provided in a wax matrix that is not absorbed but is excreted in the feces (Brunton et al, 2010). Patients may become concerned about the appearance of this substance in the feces unless the clinician mentions this. Few drugs easily penetrate intact skin. Lipid-soluble drugs may not be absorbed because the skin acts as a lipid barrier. However, other types of solutions may be absorbed. Skin that is not intact will absorb drugs more readily. Thus, it is important to apply topical medications to intact, healthy skin to ensure the correct absorption. Mucous membranes also readily absorb drugs more readily than intact skin because of increased vascularity. Topical administration has the same advantage of avoiding the first-pass metabolism of the drug through the liver (Brunton et al, 2010). Medications that cannot be given orally often can be given rectally. Rectal medications usually are given when the patient is vomiting or unconscious. One advantage is that less first-pass metabolism of the drug through the liver occurs for rectal than for oral preparations. However, rectal absorption tends to be more inconsistent and less complete than oral absorption, and some drugs may cause rectal irritation. It is not always necessary to have a specially formulated rectal preparation (Edmunds, 2013). For example, oral timed-release morphine tablets will be absorbed rectally and are useful when the patient is unable to take an occasional oral dose of pain medicine. Circulation at the site of administration is important in the drug absorption process. Decreased circulation (as seen in congestive heart failure) will result in decreased drug absorption (Katzung et al, 2011). For example, insulin injected into a thigh muscle, followed by exercise, will produce more rapid absorption of the insulin than occurs without exercise. When drugs are absorbed, they pass through cells, not between them; therefore, the drug must pass through the cellular wall or membrane. The structure of the cell membrane influences this process (Figure 3-2). The cell membrane is composed of a two-molecule layer of lipids that contains protein molecules between the lipids. It also contains carbohydrate molecules that are attached to the outer surface of the membrane. The proteins can be integral (in which case they go through the membrane) or peripheral (in which case they are attached to the surface of the membrane) (McCance & Huether, 2009). Integral proteins act as structural channels for the transportation of water-soluble substances (ions) or as carrier proteins in active transport. Peripheral proteins are enzymes. Glycoproteins may be antigenic sites in immune reactions or drug receptors. The pores permit the passage of small water-soluble substances such as water, electrolytes, urea, and alcohol (Brunton et al, 2010). Drugs cross membranes via passive diffusion or active transport. Passive diffusion involves the random movement of drug molecules from high to low concentrations. Active transport moves molecules that are moderately sized, water soluble, or ionic across cell membranes. In active transport, these molecules form complexes with carriers for transport through the membrane and then dissociate from them. Active transport requires expenditure of energy and can occur against the concentration gradient (Stringer, 2005) (Figure 3-3). In passive diffusion, the drug molecule penetrates along a concentration gradient as a result of its solubility in the lipid layer of the membrane (Figure 3-4). This transfer occurs in proportion to the magnitude of the concentration gradient across the membrane. The higher the concentration, the more rapid the diffusion across the membrane. If the drug is not an electrolyte, a steady state is attained when the concentration of free drug is the same on both sides of the membrane. If the drug is an ionic compound, the steady-state concentration will depend on the difference in pH across the membrane (Brunton et al, 2010). Whether the drug is lipophilic or hydrophilic affects absorption across cell membranes. Lipids pass through membranes better than hydrophilic molecules do. However, most cell membranes are permeable to water by diffusion or by hydrostatic or osmotic differences across the membrane (Stringer, 2005). The water may carry with it small water-soluble substances such as urea. The pH of a drug also affects diffusion. Nonionized drugs are more lipid soluble and may readily diffuse across cell membranes. Ionized drugs are lipid insoluble and nondiffusible. Acidic drugs such as aspirin become nonionized in the acidic environment of the stomach and thus can diffuse across the membranes (DiPiro et al, 2011; Stringer, 2005). A change in the acidity of the stomach, as occurs with antacids, will affect absorption of drugs. Basic drugs such as alkaloids ionize in the stomach and are not well absorbed. These drugs are better absorbed in a less acidic environment such as the small intestine. • ABC class pumps transport small molecules across membranes. They consist of two transmembrane domains and two ATP binding domains. ABC pumps are involved in the transport of small molecules, phospholipids, and lipophilic drugs in mammalian cells. In bacteria they transport amino acids, sugars, and peptides. • P-class pumps use ATP to transport ions against a gradient. They are phosphorylated during transport, which is different from the other classes of active transport pumps. Some examples of P-class pumps are the sodium-potassium pump, calcium transport in muscle cells, and the hydrogen-potassium pump in the apical membrane of the stomach. • The AV-class proton pump moves protons from one side of a membrane to the other and uses ATP as the source of energy. V-class proton pumps are a type of ATPase. They use the energy released by the hydrolysis of ATP to move protons against their concentration gradient. • F-class proton pumps have also been identified and are the subject of new research. Distribution is the transport of a drug in body fluids from the bloodstream (at the site of absorption) to various tissues in the body. The pattern of distribution depends on the pharmacokinetic activity of different types of tissue and the different physicochemical properties of drugs (Brunton et al, 2010). Drugs vary in their ability to move into various body compartments (e.g., brain, fat, lung, eye). The best way to know how much of a given drug gets into a particular body compartment is to consult standard reference texts. Because blood is an easily accessible body fluid, blood concentrations often are studied to determine how they relate to drug concentrations in other body compartments. The dose-related effects of a drug are then correlated with a given blood concentration or range of concentration. Once a relationship has been established, blood concentrations can be used to monitor therapy. Distribution consists of two phases. The first involves movement from the site of administration into the bloodstream. Delivery of the drug into the tissue at the site of action is the second phase of distribution (Katzung et al, 2011). The volume of distribution (Vd) is a concept that is useful when one seeks to understand where the drug goes once it is absorbed. Volume of distribution is a description of the amount of space into which a drug can be spread or distributed. It is the calculated volume or size of a compartment necessary to account for the total amount of drug in the body if it were present throughout the body at the same concentration found in the plasma (Brunton et al, 2010). Because most drugs are not equally distributed, this is a theoretical concept and not a real volume. It is, however, useful in predicting drug concentrations, understanding how well a drug is absorbed into tissues, and understanding whether it will be accumulated in the tissues. If a drug has a small volume of distribution, it stays in the central compartment and is not widely distributed. If the drug has a large volume of distribution, it is found widely throughout the body. The larger the Vd, the more drug there is in the tissue. Because we are able to measure the amount of drug in the bloodstream only through serum levels, calculation of the volume of distribution allows us to estimate or predict the concentration of drug in the tissue (Stringer, 2005). The Vd may be calculated by examining the following relationships: then This means that the Vd is equal to the dose of drug in the body divided by the concentration in the blood (Brunton et al, 2010). Water-soluble drugs have a Vd that is similar to the plasma volume because they are distributed into the blood. The plasma volume for a normal adult is about 3 to 5 liters. Lipophilic drugs have a larger Vd. The total fluid volume in the body is about 40 liters for a 70-kg (150-lb) person. Because the Vd of a lipophilic drug can exceed this volume, it is important to remember that this calculation is of a hypothetical volume, not a real volume. It is the volume that would be required to contain the entire drug in the body if the drug were distributed in the same concentration as in the blood or plasma (Brunton et al, 2010). Drugs that are highly protein bound (>90%) have a volume of distribution that is about the same as the amount of plasma. This is a small volume of distribution. An example would be the thyroid hormones. If the patient has a decreased serum albumin, more active drug is available for protein-bound drugs. If two protein-bound drugs are used, the most tightly protein-bound drug will tend to displace the other. Examples of these drugs include furosemide and the cephalosporins (Brunton et al, 2010; DiPiro et al, 2011). Another common change that may affect the distribution of drugs in older adults is a decrease in serum albumin. Albumin concentrations decrease slightly with age in most elderly patients, although significant changes that may affect drug therapy may be seen in the chronically ill or malnourished elderly patient (Bourne, 2007). Albumin is the most common protein that binds to various acidic drugs. Significant decreases in albumin may result in a greater free concentration of highly protein-bound drugs. Box 4-3 lists some drugs that have significant protein binding, which may result in greater free concentrations when albumin is significantly reduced. Generally, drugs that are highly protein bound to albumin should be prescribed in reduced doses for patients with low serum albumin values (Bourne, 2007). A practical example of the clinical significance of this relationship can be described with the anticonvulsant phenytoin. In an elderly patient with a low serum albumin concentration (normal, 3.5 to 5 mg/dl), the phenytoin level reported from virtually all laboratories is the bound concentration. In a hypoalbuminemic individual, this value may appear normal or even subtherapeutic. This is because of the greater amounts of “free” or nonbound phenytoin that are getting into the tissue and acting at the receptor level but are not portrayed in the total serum level. The actual level may be much higher or even in the toxic range. Treatment decisions with older adults should not be based solely on drug levels. Treatment decisions must be based on consideration of both patient characteristics and drug levels. Other protein changes also may have an influence on drug therapy. Patients with acute disease—such as myocardial infarction, respiratory distress, or infectious insult, for example—may experience increases in α1-glycoprotein, an acute phase reactant protein. This may result in the increased binding of weakly basic drugs, including propranolol or lidocaine, and a less-than-normal response to therapy. Data are lacking concerning the real significance of changes in α1-glycoprotein and drug therapy in the elderly (DiPiro et al, 2011). The distribution of drugs from the bloodstream to the central nervous system is different than that through other cell membranes. The endothelial cells of the brain capillaries do not have intercellular pores and vesicles. Passive distribution of hydrophilic drugs is restricted. However, lipophilic drugs will easily pass the blood-brain barrier, limited only by cerebral blood flow (Brunton et al, 2010). Highly fat-soluble drugs also cross the blood-brain barrier easily and are likely to cause central nervous system side effects such as confusion and drowsiness. Biotransformation is the chemical inactivation of a drug through conversion to a more water-soluble compound that can be excreted from the body. Biotransformation occurs primarily in the liver, but it also may be found in the lungs and in the GI tract. It involves two major steps in enzyme activity. Phase I makes the drug more hydrophilic through oxidation, reduction, or hydrolysis. Minor changes in the structure of the drug make it more hydrophilic but allow it to maintain all or part of its pharmacologic activity. The cytochrome P450 enzyme system is a part of phase I. Phase II is called glucuronidation. It involves conjugation, or attachment of particles to the molecule, making it a highly water-soluble substance with little or no pharmacologic activity (Brunton et al, 2010). If blood flow to the liver is decreased, drugs will be metabolized more slowly, leading to a longer duration of action. Lipophilic drugs pass easily through membranes, including renal tubules, making them difficult to excrete. Biotransformation changes a lipophilic drug that is active and transforms it into a hydrophilic inactive compound that is readily excreted. However, metabolites occasionally have biologic activity or toxic properties (Figure 3-5). The cytochrome P450 enzyme system operates throughout the body. It is concentrated in the liver, intestine, and lungs. The P450 enzyme system resides in the ribosomes, which are sacs in the endoplasmic reticulum. It is named P450 because this is the length of the wave of light that these enzymes absorb. Chemically, the enzyme is a glycoprotein or a sugar plus a protein. Many of the proteins contain heme—hence the name chrome. This family of enzymes is divided into groups according to similarity. At least 40 major groups have been identified in humans so far. The major groups named 1, 2, 3, and 4 are known to be involved in drug interactions. These major groups are further divided into groups by their chemical structure, named A, B, and C. The A, B, and C groups are then divided into subgroups named 1, 2, 3, and so on. The groups that are most important in human drug interactions are CYP 2D6, CYP 3A3/4, CYP 1A2, CYP 2C9/10, and CYP 2C19. In the groups with 3/4 and 9/10, the two are so close in structure that they are difficult to differentiate and have very similar actions (Dresser et al, 2000; Lim et al, 2005; Zhou et al, 2004, 2005). The essential facts to master about the P450 enzyme system are that six primary enzymes account for the metabolism of nearly all clinically important drugs, and two of these systems are critically important for drug metabolism (Table 3-1). TABLE 3-1 Examples of Clinically Relevant Drugs Metabolized by Various CYP Enzymes From Flockhart D: Cytochrome P450 drug-interaction table, Division of Clinical Pharmacology, Indiana University Department of Medicine, 2007 (Available at http://medicine.iupui.edu/flockhart/table.htm). Of the other four enzymes, the most notable features of each are as follows: • CYP 2C19—metabolizes proton pump inhibitors, NSAIDs, and β-blockers • CYP 2C9—metabolizes sulfonylureas, NSAIDs, (S)-warfarin, and sildenafil citrate (Viagra) • CYP 1A2—metabolizes acetaminophen, (R)-warfarin, theophylline, caffeine, diazepam (Valium), and verapamil • CYP 2E1—metabolizes acetaminophen, ethanol, inactivation of toxins, and dextromethorphan A drug can be a substrate or one that is affected by alteration of its enzyme metabolism. A drug can also be the one that causes the alteration in the enzyme metabolism of another drug by being an inhibitor or an inducer. The cytochrome P450 enzyme system may speed up a reaction because it causes the drug to change to a more hydrophilic substance. Any drug that causes the enzyme to metabolize more slowly or decreases the capacity of the enzyme pathway is called an inhibitor. For example, if a patient on fluoxetine (Prozac) takes warfarin, the fluoxetine inhibits the P450 enzyme system from metabolizing warfarin and may produce an exaggerated therapeutic response (bleeding). A drug that causes the enzymes to metabolize the substrate more quickly is called an inducer. These types of drugs increase enzyme activity by increasing the number of CYP 450 enzymes. Any drug can be involved in this process in two ways. It can be the drug or substrate that is being acted upon, or it can be the inhibitor or inducer that is acting on the enzyme to increase or decrease enzyme conversion of the substrate drug into an inactive compound. The same drug can be both a substrate and an inducer or inhibitor (Brunton et al, 2010). For example, carbamazepine is an auto inducer—it induces its own metabolism. It is the enzyme system and not the drug that is being induced or inhibited (see Figure 3-5 to examine these relationships). A drug can inhibit an enzyme pathway through two mechanisms. The first is competition, which is not usually a problem. If it occurs, it occurs immediately. Most inhibition is metabolic. The inhibitor drug decreases the production of the enzyme. It shrinks the enzyme pathway. This is not an immediate reaction; it may take anywhere from 24 hours up to a week to see the effect, depending on the half-life of the drug (Brunton et al, 2010). From a pharmacokinetic standpoint, the major effects of drug–drug interactions are understood in terms of causing a high or low plasma and tissue level of the drug. In addition to cytochrome P450, oxidation of drugs and other xenobiotics can be mediated by non-P450 enzymes, the most significant of which are flavin monooxygenase, monoamine oxidase, alcohol dehydrogenase, aldehyde dehydrogenase, aldehyde oxidase, and xanthine oxidase. Drug oxidation catalyzed by some of these enzymes may often produce the same metabolites as those generated by P450; thus, drug interactions may be difficult to predict without a clear knowledge of the underlying enzymology. Although oxidation catalyzed by non-P450 enzymes can lead to drug inactivation, oxidation may be essential for the generation of active metabolites that create drug action (Brunton et al, 2010). Phase II or biotransformation consists of conjugate reactions in which a compound is added to the drug. These reactions bind a chemical group to the drug compound via a covalent linkage. The chemical groups added to the drug are generally highly polar, or ionized. This makes them water soluble and generally inactive. However, a few of these conjugate compounds are active (Brunton et al, 2010). When a medication is taken orally, it passes from the intestine directly to the liver by way of the hepatic portal blood flow (via the portal vein). Many drugs, such as nitroglycerin and estrogen, are extensively metabolized to inert compounds when they first pass through the liver. Because of this, very large amounts of the drug must be given for a sufficient dose to remain after the first pass through the liver. This is known as the first-pass effect (Stringer, 2005). It is why these drugs are often given via an alternative route, such as sublingual or topical, to avoid the liver on the first pass. Alternative route dosing also makes it possible to give a smaller amount of the drug and have it be effective.
General Pharmacokinetic and Pharmacodynamic Principles
Drug Nomenclature
Pharmacokinetics
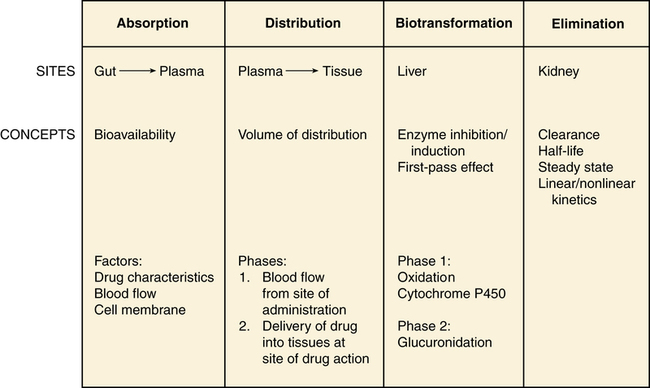
Absorption
Factors That Affect Absorption
Drug Characteristics
Routes of Administration
Oral
Topical
Rectal
Blood Flow
Cell Membrane Characteristics
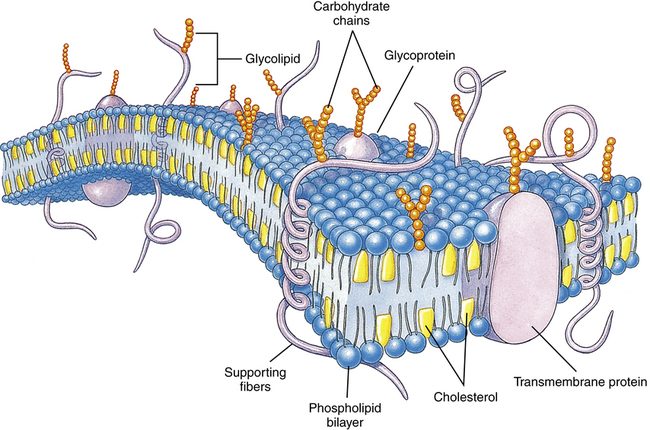
The lipid bilayer provides the basic structure and serves as a relatively impermeable barrier to most water-soluble molecules. (Modified from Thibodeau GA, Patton KI: Structure and function of the human body, ed 13, St Louis, 2008, Mosby.)
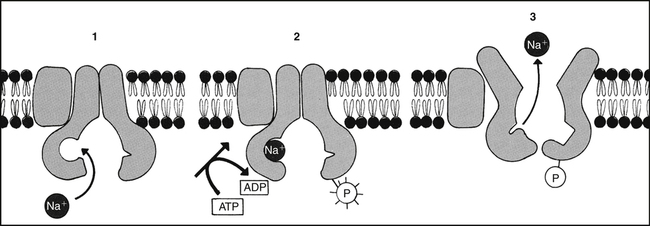
Metabolic energy is necessary for the active transport of many substances, including Na. (Modified from Alberts B et al, editors: Molecular biology of the cell, ed 3, New York, 1994, Garland.)
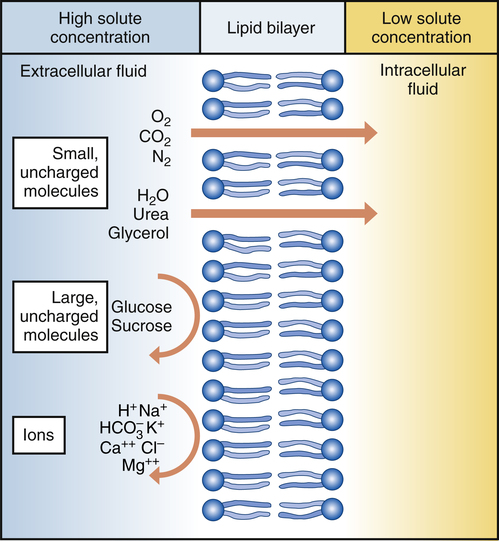
Oxygen, nitrogen, water, urea, glycerol, and carbon dioxide can diffuse readily down the concentration gradient. Macromolecules are too large to diffuse through pores in the plasma membrane. Ions may be repelled if the pores contain substances with identical charges. (From Thibodeau GA, Patton KI: Understanding pathophysiology, ed 6, St Louis, 2008, Mosby.)
Distribution
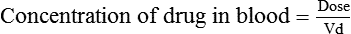
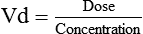
Biotransformation (Metabolism)
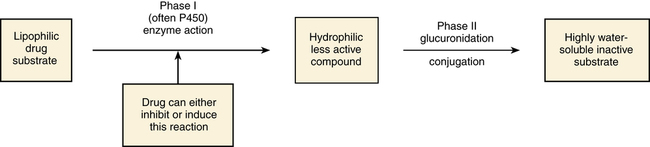
Phase I: Oxidation or Reduction of Drugs
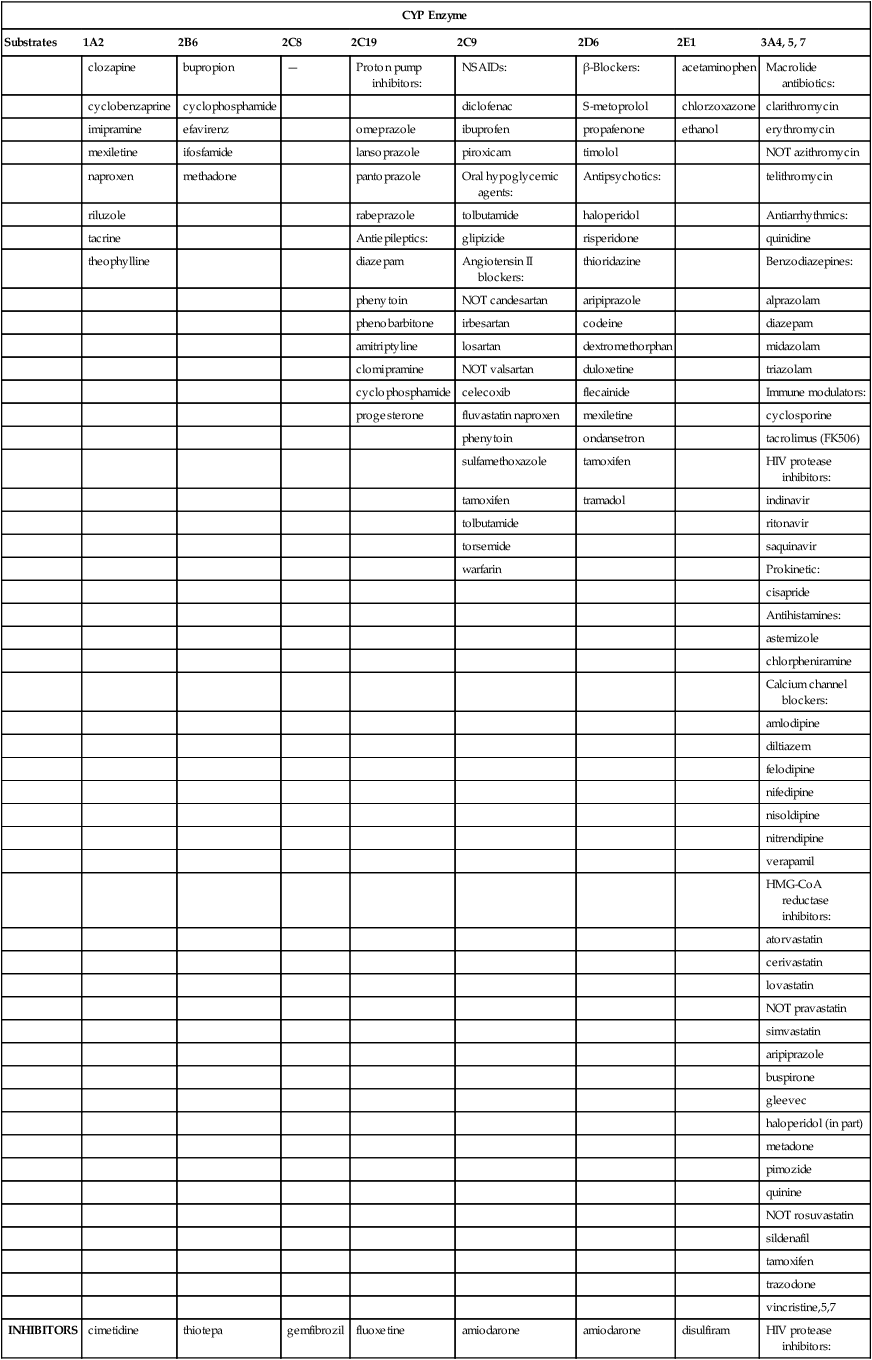
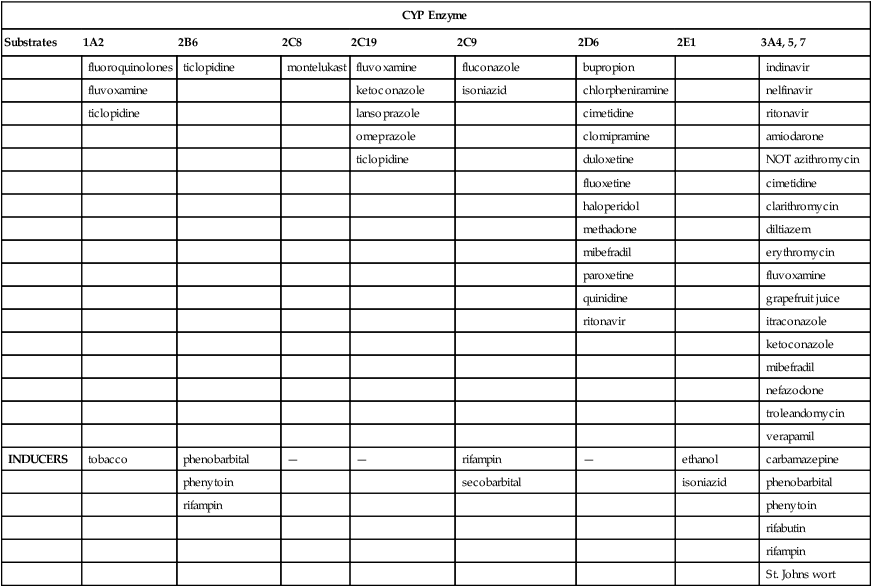
CYP Enzyme
Substrates
1A2
2B6
2C8
2C19
2C9
2D6
2E1
3A4, 5, 7
clozapine
bupropion
—
Proton pump inhibitors:
NSAIDs:
β-Blockers:
acetaminophen
Macrolide antibiotics:
cyclobenzaprine
cyclophosphamide
diclofenac
S-metoprolol
chlorzoxazone
clarithromycin
imipramine
efavirenz
omeprazole
ibuprofen
propafenone
ethanol
erythromycin
mexiletine
ifosfamide
lansoprazole
piroxicam
timolol
NOT azithromycin
naproxen
methadone
pantoprazole
Oral hypoglycemic agents:
Antipsychotics:
telithromycin
riluzole
rabeprazole
tolbutamide
haloperidol
Antiarrhythmics:
tacrine
Antiepileptics:
glipizide
risperidone
quinidine
theophylline
diazepam
Angiotensin II blockers:
thioridazine
Benzodiazepines:
phenytoin
NOT candesartan
aripiprazole
alprazolam
phenobarbitone
irbesartan
codeine
diazepam
amitriptyline
losartan
dextromethorphan
midazolam
clomipramine
NOT valsartan
duloxetine
triazolam
cyclophosphamide
celecoxib
flecainide
Immune modulators:
progesterone
fluvastatin naproxen
mexiletine
cyclosporine
phenytoin
ondansetron
tacrolimus (FK506)
sulfamethoxazole
tamoxifen
HIV protease inhibitors:
tamoxifen
tramadol
indinavir
tolbutamide
ritonavir
torsemide
saquinavir
warfarin
Prokinetic:
cisapride
Antihistamines:
astemizole
chlorpheniramine
Calcium channel blockers:
amlodipine
diltiazem
felodipine
nifedipine
nisoldipine
nitrendipine
verapamil
HMG-CoA reductase inhibitors:
atorvastatin
cerivastatin
lovastatin
NOT pravastatin
simvastatin
aripiprazole
buspirone
gleevec
haloperidol (in part)
metadone
pimozide
quinine
NOT rosuvastatin
sildenafil
tamoxifen
trazodone
vincristine,5,7
INHIBITORS
cimetidine
thiotepa
gemfibrozil
fluoxetine
amiodarone
amiodarone
disulfiram
HIV protease inhibitors:
fluoroquinolones
ticlopidine
montelukast
fluvoxamine
fluconazole
bupropion
indinavir
fluvoxamine
ketoconazole
isoniazid
chlorpheniramine
nelfinavir
ticlopidine
lansoprazole
cimetidine
ritonavir
omeprazole
clomipramine
amiodarone
ticlopidine
duloxetine
NOT azithromycin
fluoxetine
cimetidine
haloperidol
clarithromycin
methadone
diltiazem
mibefradil
erythromycin
paroxetine
fluvoxamine
quinidine
grapefruit juice
ritonavir
itraconazole
ketoconazole
mibefradil
nefazodone
troleandomycin
verapamil
INDUCERS
tobacco
phenobarbital
—
—
rifampin
—
ethanol
carbamazepine
phenytoin
secobarbital
isoniazid
phenobarbital
rifampin
phenytoin
rifabutin
rifampin
St. Johns wort
Phase II: Biotransformation
First-Pass Effect
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


General Pharmacokinetic and Pharmacodynamic Principles

Only gold members can continue reading. Log In or Register to continue
