Overview of the Problem
Cancer is a disease of cells that escape the control mechanisms of orderly cell growth and acquire the ability to proliferate, invade normal tissues, and metastasize. It is generally assumed that cancer is a
clonal disorder derived from a single transformed cell (see below). The fundamental research issue was to determine whether cancer was the result of stimulation of cell growth, damage to the mechanisms regulating normal cell replication, or both.
Marx (1986) referred to this dilemma as the Yin and Yang of cell growth control, referring to the old Chinese concept of contradictory forces in nature.
There were several significant problems with the study of molecular events in cancer. One of them was the
heterogeneity of cancer cells—the observation that few, if any, cancer cells were identical. This phenomenon of cancer cell diversity was extensively studied by
Fidler et al (1982,
1985), who documented that, in experimental tumors in mice, some cancer cells were capable of forming metastases and others were not. It has also been known for several years that the number and type of chromosomal abnormalities increased with the progression of cancer, reflecting the genomic instability in the cancer cells (recent review in
Kiberts and Marx, 2002).
Nowell (1976), who studied this phenomenon in leukemia, called it
clonal evolution. In cytogenetic studies of fully developed solid cancers, the number of chromosomes in individual cancer cells is often variable and other aberrations of chromosomes may also occur (see
Chap. 4). It is not an exaggeration to state that advanced human cancer represents a
state of genetic chaos. The diversity of cancer cells, even within the same tumor, made it very difficult to assess whether observed molecular genetic abnormalities had universal significance or were merely an incidental single event (recent reviews in
Tomlison et al, 2000, and
Hahn and Weinberg, 2002).
The type of material that was available to the basic science investigators also posed similar problems. Fragments of cancerous tissues available for such purposes were usually derived from advanced tumors that were likely to show a great deal of heterogeneity and genetic disarray. In vitro
culture of human cancers is technically difficult, and the cell lines derived therefrom usually represented a single clone of cells that is not necessarily representative of the primary tumor.
Further complications arose when DNA or RNA were extracted from such tissue samples for molecular analysis. Besides tumor cells, such tissues always contained an admixture of benign cells from blood vessels, connective tissue stroma, inflammatory cells, and remnants of the normal organ of origin. The question as to what constituted tumorspecific findings, rather than findings attributable to normal cells, was often difficult to resolve. Many of these difficulties persist.
Some solutions to these dilemmas came from several unrelated sources. One of them was the discovery of growthpromoting DNA sequences, known as oncogenes, and their precursor molecules, the protooncogenes, in an experimental system of transformed rodent cells. The protooncogenes and oncogenes could be isolated and sequenced. The search could now begin for matching sequences in the DNA extracted from normal human tissues and cancer. The protooncogenes and oncogenes and their role in cancer are described below.
Another breakthrough occurred with the study of the patterns of occurrence of
retinoblastoma, an uncommon malignant tumor of the retina in children.
Knudson (1971) anticipated that a fundamental genetic abnormality accounted for the familial pattern of this disease. This abnormality was subsequently identified as a deficiency or absence of a gene located on chromosome 13, which was named the
retinoblastoma (Rb) gene (see below for further discussion). Similar studies of
families with “cancer syndromes” were also conducted. Such families, described by a number of investigators (
Gardner, 1962;
Li and Fraumeni, 1969; summaries in
Lynch and Lynch, 1993;
Fearon, 1997;
Varley et al, 1997;
Frank 2001) were characterized by a high frequency of occurrence of cancers in various organs. The most important cancer syndromes are listed in
Table 7-2. By a variety of techniques known as
linkage analysis, the genetic abnormalities could be identified and the genes localized—first to chromosomes, then to segments of chromosomes, and, finally, to the specific location on the affected chromosome. The isolation and sequencing of such genes were an essential step in studying their function and interaction with other genes.
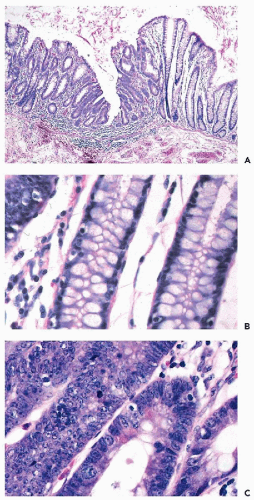
Of special value in this research were
families with congenital polyposis of the colon, a disease process in which the patients develop innumerable benign colonic polyps and, unless treated, invasive cancer of the colon sooner or later. A group at The Johns Hopkins medical institutions in Baltimore, MD, led by Vogelstein, Fearon, and others, undertook a systematic study of genetic changes occurring
in benign colonic polyps, polyps with atypical features, early cancer, and invasive colonic cancer. These studies led to a
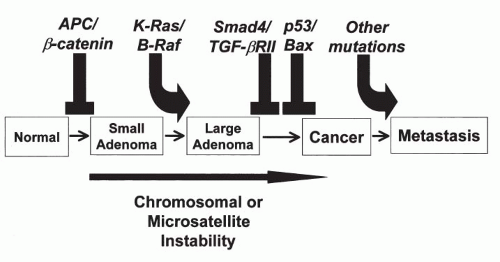
model of carcinogenesis in the colon that postulated a sequence of genetic abnormalities leading from normal epithelium to polyps to cancer (
Fig. 7-4). Although this model is not likely to be applicable to all cancers of the colon, let alone other organs, it stimulated a great deal of research on carcinogenesis. Perhaps the most important developments, resulting directly or indirectly from the studies of familial cancer, were the discovery of the role played by regulatory genes
(tumor suppressor genes) in the events of cell cycle and the
relationship of genes involved in cancer genesis with adhesion molecules that regulate the relationship of cells to each other and to the underlying stroma. These observations are discussed below.
Another development that proved to be of significance in this research was the Human Genome Project, which provided a great deal of information on the distribution of genes on human chromosomes. Although the map of the human genome has been completed and the significance and role played by most of the genes remain unknown, commercial probes to many of these genes have become available that allow the study of genetic abnormalities in various human cancers. The emerging information is, unfortunately, enormously complex and so far has shed little light on the initial events, or sequence of events, in solid human cancer. Still, the genome project led to the discovery of the human breast cancer genes BRCA1 and BRCA2, to be discussed below.
Protooncogenes and Oncogenes
The first significant observation shedding light on the molecular mechanisms of cancer was the discovery of
oncogenes in the 1980s (summary in
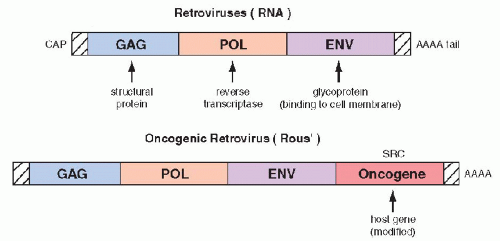
Bishop, 1987). The oncogenes were first identified in experimental systems in which cultured, benign rodent cells were infected with
oncogenic RNA viruses (retroviruses) and were transformed into cells with malignant features. The viral RNA, by means of the enzyme reverse transcriptase is capable of producing cDNA that is incorporated into the native DNA (genome) of the cell, which becomes the source of viral replication. It has been observed that
regulatory genes of host DNA, named protooncogenes, which may be incidentally appropriated by the viral genome,
are essential in the transformation of the infected cells into cells with malignant features. The “stolen”
host cell genes, when either
overexpressed or modified (mutated),
become a growth-promoting factor that has been named an oncogene (
Fig. 7-5). The first oncogenes discovered were named
ras (
retrovirus-
associated
sarcoma or
rat
sarcoma). Several variants of the
ras oncogenes were subsequently discovered and described with various prefixes, such as
Ki–
ras, Ha–
ras, and
N–
ras, reflecting the initials of the investigators.
Shortly after the discovery of the first protooncogenes and oncogenes and their sequencing, their presence could be documented by Southern blotting and similar techniques in DNA from normal human tissues, in human tumors, and in cell lines derived therefrom. On the assumption that the study of oncogenes will provide the clue to the secrets
of abnormal cell proliferation in cancer, the search for other oncogenes and growth-promoting factors began in earnest and led to the discovery of a large number of such genes that have now been sequenced and traced to their chromosomal sites.
Two fundamental modes of oncogene function have been identified—
overexpression (amplification) of a normal protooncogene product, and a
point mutation, a single nucleotide change in an exon of the gene, leading to a modified protein product. It is known that some oncogenes can be activated because their original chromosomal site has been disturbed by breakage and
translocation of chromosomal segments, as observed in lymphomas and leukemias (see below). They may also be overexpressed in chromosomal fragments, such as C-
myc oncogene, observed in the
double-minute chromosomes of neuroblastoma (see
Chap. 4).
The protooncogenes and the oncogenes exercise their activity through their protein products, many of which have been identified. For example, the genes of the ras family encode a group of proteins of 21,000 daltons, known as p21. Contrary to the initial hopes that all oncogenes would have a simple, well-defined function in the transformation of benign into malignant cells, it is now evident that the oncogenes are a diverse family of genes, with different locations within the cell and different functions. Several oncogenes have been traced to the nucleus (e.g., myc, myb, fos, jun), presumably interacting directly with DNA. Other proteins encoded by oncogenes have an affinity for cell membranes (e.g., ras, src, neu) or the cytoplasm (e.g., mos). These latter two groups of oncogenes appear to interact, on the one hand, with cytoplasmic and cell membrane receptors and, on the other hand, with enzymes, such as tyrosine kinase, that play a role in DNA replication. It is possible that the oncogenes located on cell membranes are instrumental in capturing circulating growth factors that stimulate proliferation of cells.
In
solid human tumors, the activation or overexpression of various oncogenes has been shown to be a
common event, unlikely to establish a simple cause-effect relationship between oncogene activation and the occurrence of human cancer. The presence of oncogene products could be demonstrated either by molecular biology techniques or by immunocytochemistry in many different human cancers. As an example, the presence of the
ras oncogene product, p21, has been documented by us and others in gastric, colonic, and mammary cancer cells, and in several other human tumors (
Czerniak et al, 1989,
1990,
1992). In cytochemical studies, it was noted that oncogene products are
variably expressed by cancer cells, some of which stain strongly and some that do not stain at all, suggesting heterogeneity of oncogene expression. It is possible that the expression of the oncogene products is, to some extent, cell cycle dependent (
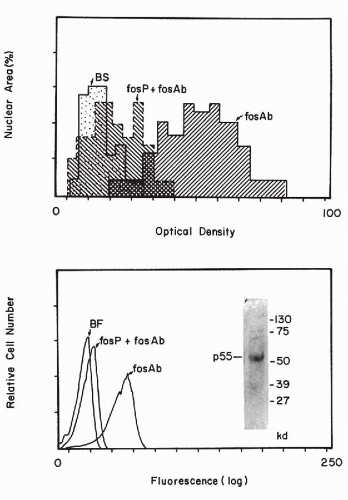
Czerniak et al, 1987). With image analysis and flow cytometric techniques (see
Chaps. 46 and
47), the amount of the reaction product can be measured (
Fig. 7-6).
Press et al (1993) stressed that immunocytologic microscopic techniques with specific antibodies are probably more reliable in assessing the expression of an oncogene in tissues than is the Southern or northern blotting technique. The blotting techniques require the destruction of the tissue samples and, therefore, fail to provide information on the makeup of the destroyed tissue and on the proportion of normal cells in the sample.
However, there is no agreement on the diagnostic or prognostic value of such measurements in human solid tumors, with a few exceptions. For example, the elevated expression of the product of the
oncogene HER2 (also known as
c-erbB2), a transmembrane receptor protein, indicates poor prognosis and rapid progression of breast cancers in about 25% of affected women (
Slamon et al, 1989). In fact, an
antibody to the protein product of this gene has been developed commercially for human use and is of benefit in prolonging life in some women with advanced metastatic breast cancer (see
Chap. 29). This is one of the first indications that knowledge of the oncogenes or tumorpromoting factors may be of benefit to patients. Although oncogenes play an important role in human cancer, their precise role is complex (summary in
Krontiris, 1995).
Weinstein (2002) suggested that individual cancers are “addicted” to their specific oncogenes and suggested that oncogene suppression may lead to cure.
As on example, the drug Gleevac (Novarrtis) has been shown to be effective against chronic myclogenous leukemia by blocking the oncogenic protein bcr = abl, the product of chromosome translocation.
Tumor Suppressor Genes and Gatekeeper Genes
The oncogene story became even more complicated with the identification of genes known collectively as tumor suppressor genes or gatekeeper genes. As previously mentioned, this research has been stimulated by studies of
families with cancer syndromes (recent summary in
Fearon, 1997; see
Table 7-2). The first such gene discovered was the
retinoblastoma (Rb) gene, located on the short arm of chromosome 13. Retinoblastoma is an uncommon, highly malignant eye tumor of childhood that occurs in two forms: (1) a familial form, in which usually both eyes are affected, and (2) a sporadic form, in which one eye is affected. Following treatment of retinoblastoma, other cancers, such as osteogenic sarcoma, may develop in the affected children. Thus, the defect of the Rb gene may have multiple manifestations.
It was postulated by
Knudson in 1971 that retinoblastomas are the consequence of two mutational events
(two-hit theory of cancer). The familial form of retinoblastoma implied a hereditary defect of some sort, supplemented by a single additional sporadic mutation, leading to cancer. In the sporadic form, two mutational events were anticipated against a normal genetic background. In retinoblastoma, the gene on chromosome 13 was frequently deficient or absent, thus fulfilling the first requirement of Knudson’s hypothesis. This gene has now been sequenced and its anti-tumor activity has been confirmed in vitro by
Huang et al in 1988. It has been learned in recent years that the protein
product of the Rb gene regulates the expression of one of the proteins regulating the cell cycle, known as
D cyclins, which govern the transition of cells from G
0 to G
1 stage of
mitosis. It is postulated that
the absence of, or damage to, the Rb gene deregulates the cell cycle, leading to cancer.Another important regulatory gene is
p53, a protein product of the gene located on the short arm of the chromosome 17 (
Levine et al, 1991). p53 is a DNA binding protein that
regulates the transcription of DNA, its repair by a cascade of other proteins, and is, therefore, considered to be a
“guardian of the genome” (
Lane, 1992). If a transcriptional error occurs, the replication is stopped until the error is repaired. The mechanism of arrest is mediated by a cell cycle inhibitor, protein p21
WAF1/CIP1, which is different from the p21 protein of the
ras gene. If the repair is not executed, the cell may enter into the cycle of programmed cell death or
apoptosis, discussed in detail in
Chapter 6.
The natural p53 product is short-lived and difficult to demonstrate; however, a gene mutation leads to a modified protein that has a much longer life span and can be demonstrated by a variety of techniques, including immunocytochemistry.
Loss of heterozygosity of p53 (inactivation or mutation of one of the two identical genes within the cell) is a very
common event in many human cancers of various organs, mainly in advanced stages (see later text). However, in some cancers, such as high-grade cancer of the endometrium, the mutation of p53 is presumed to occur as an early event (see
Chap. 13). The presence of mutations of the Rb gene and of the p53 protein has been shown to confer a poor prognosis on some cancers, such as cancers of the bladder (
Esrig et al, 1993;
Sarkis et al, 1993), some malignant lymphomas (
Ichikawa et al, 1997), and chondrosarcomas (
Oshiro et al, 1998).
Other tumor suppressor genes include the recently identified
breast cancer genes, BRCA1 and BRCA2 (see
Table 7-2). The mutations of these genes have been observed in a larger proportion of Jewesses of Eastern European (Ashkenazi) origin than in other comparable groups of women (recent summary in
Hofmann and Schlag, 2000). Although some of these women are at an increased risk for breast, and, to a lesser extent, ovarian and tubal cancer, and deserve close follow-up, the extent of risk for any individual patient cannot be assessed. In some of these women, preventive measures, such as a prophylactic mastectomy and oophorectomy have been proposed (
Schraq et al, 1997). Clearly, many such dilemmas will occur as new risk factors for cancer are discovered. Silencing of tumor suppressor genes may be caused by
methylation that does not involve DNA mutations (recent summary in
Herman and Baylin, 2003).
Another set of genes involved in malignant transformation of normal cells into cancer cells is the
susceptibility genes, considered by
Kinzer and Vogelstein (1998) as “
caretakers of the genome.” These genes, when mutated or inactivated, contribute indirectly to the neoplastic process, probably by regulating the relationship of the transformed cells to connective tissue stroma. Such genes have been observed in a colon cancer syndrome known as the
hereditary nonpolyposis colorectal cancer (summary in
Kinzer and Vogelstein, 1996). These observations bring into focus another critical issue in reference to cancer, namely
the relationship of cancer cells to adhesion molecules that normally maintain order within the tissue and are critical in understanding the mechanism of cancer invasion and metastases. Several such molecules, such as
cadherins (
Takichi, 1991),
integrins (
Albelda, 1993),
lamins (
Liotta et al, 1984), and
CD44 (
Tarin, 1993), have been studied and have been shown to be of significance in cancer invasion and metastases.
It is the consensus of most investigators that cancer is a multistep process that includes sequential and progressive accumulation of oncogenes and inactivation of growth-regulating genes.
Gene Rearrangement in Malignant Lymphomas and Leukemias: Effects of Translocations
Chromosomal abnormalities in leukemias have been studied since the onset of contemporary genetics. The
Philadelphia chromosome (Ph), a shortened chromosome 22, described by
Nowell and Hungerford in 1960 in chronic myelogenous leukemia, was the first documented chromosomal abnormality characteristic of any human cancer (see
Chap. 4). With the availability of the techniques of chromosomal banding and molecular biology, the genetic changes in this group of diseases could be studied further. Many of these fundamental observations are of diagnostic and prognostic value. In many disease processes within this group of cancers, an
exchange of chromosomal segments or
translocation is observed (see
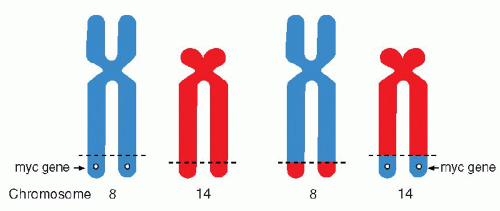
Chap. 4 for a discussion of cytogenetic changes in human cancer). Thus, it has been shown that the Ph chromosome is the result of a translocation of portions of the long arm of chromosome 22 to the long arm of chromosome 9 [abbreviated as t(q9;q22)]. In certain forms of malignant lymphoma (notably in lymphomas of Burkitt’s type), there is a reciprocal translocation between segments of chromosomes 14 and 18 (
Fig. 7-7).
The results of a translocation can be:
It is the last property that has served as a template for development of a new drug (Gleevec, Novartis) that is effective
against the product of chromosomal translocation in chronic myelogenous leukemia. The new agent also appears to be active against a group of gastrointestinal tumors known as GIST (see
Chap. 24).
Many genes affected by translocations have been localized, identified, and sequenced (
Mitelman and Mertens, 1997). It is now known that the genes involved are often related to the principal sites encoding immunoglobulin genes. Adjacent genes often encode for certain oncogenes. For example, the 14:18 chromosomal translocation in B-cell lymphomas affects a gene known as
bcl-2 and, in Burkitt’s lymphoma, the c-
myc gene. Both the bcl-2 and c-myc genes have been shown to be
inhibitors of programmed cell death or apoptosis and it is assumed that their mutation prevents apoptosis of genetically deficient cells and, thus, contributes to an unregulated proliferation of abnormal cells or cancer (
Sanchez-Garcia, 1997).
Immortality of Cancer Cells
In 1965, Hayflick pointed out that
normal cells have a limited life span and die after 50 generations. These constraints are not applicable to
cancer cells, which
are theoretically immortal, as pointed out by
Cairns (1975). Contrary to normal cells, given favorable conditions necessary for survival, cancer cells can live forever, and, in fact, they do so in tissue cultures. The reasons for the ability of cancer cells to proliferate without constraints are complex and not fully understood. One of the likely reasons is that cancer cells are deficient in control mechanisms protecting normal cells from faulty reproduction of DNA. In favor of this concept is the presence of the genetic defects, such as a
mutated p53, in some cancer cells. This heritable defect in DNA control mechanisms may explain why the initial genetic changes lead to a cascade of events that result in ever increasing molecular (and chromosomal) disorders, discussed previously.
It is also possible that the chromosomes in cancer cells have a better mechanism of survival that prevents them from entering senescence, customary in normal cells. The guilty party may be the group of enzymes known as
telomerases, enzymes governing the formation of telomeres, or the terminal endings of chromosomes (
Blackburn, 1990). In normal cells, the length of the telomeres shrinks with age, presumably preventing the chromosomes from normal replication and leading to cell death after the 50 generations observed by Hayflick.
Telomerases may be overexpressed in cancer and provide additional telomeres, thus preventing the senescence of chromosomes and leading to the immortality of cancer cells (
Haber, 1995).
Measuring the elevated expression of telomerase in cells has been used in the diagnosis of cancer (see
Chap. 26).
The observations on the role of telomeres and telomerase in normal and cancerous cells are somewhat paradoxical; longevity of cells (and, by implications, multicellular organisms) and cancers have a common denominator. It is a matter for pure speculation at this time whether the efforts at extending the span of normal human life will inevitably lead to cancer. The same reasoning may, perhaps, be applied to the efforts at reversal of the malignant process by replacing damaged genes with intact genes. Such procedures have been repeatedly and successfully performed in vitro on tissue cultures but, so far, there is no reported evidence known to us of a successful application of such a procedure to multicellular organisms in vivo. It remains to be seen what long-term consequences this sort of a genetic manipulation of complex organisms may produce.