POLYURIA
By history, it is often difficult for patients to distinguish urinary frequency (often of small volumes) from true polyuria (>3 L/d), and a quantification of volume by 24-h urine collection may be needed (Fig. 61-4). Polyuria results from two potential mechanisms: (1) excretion of nonabsorbable solutes (such as glucose) or (2) excretion of water (usually from a defect in ADH production or renal responsiveness). To distinguish a solute diuresis from a water diuresis and to determine whether the diuresis is appropriate for the clinical circumstances, urine osmolality is measured. The average person excretes between 600 and 800 mosmol of solutes per day, primarily as urea and electrolytes. If the urine output is >3 L/d and the urine is dilute (<250 mosmol/L), total mosmol excretion is normal and a water diuresis is present. This circumstance could arise from polydipsia, inadequate secretion of vasopressin (central diabetes insipidus), or failure of renal tubules to respond to vasopressin (nephrogenic diabetes insipidus). If the urine volume is >3 L/d and urine osmolality is >300 mosmol/L, a solute diuresis is clearly present and a search for the responsible solute(s) is mandatory.
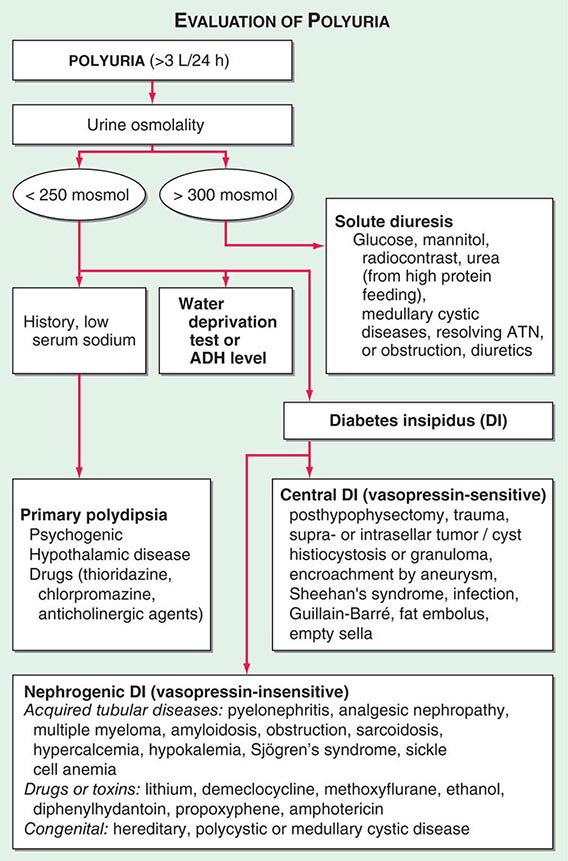
FIGURE 61-4 Approach to the patient with polyuria. ADH, antidiuretic hormone; ATN, acute tubular necrosis.
Excessive filtration of a poorly reabsorbed solute such as glucose or mannitol can depress reabsorption of NaCl and water in the proximal tubule and lead to enhanced excretion in the urine. Poorly controlled diabetes mellitus with glucosuria is the most common cause of a solute diuresis, leading to volume depletion and serum hypertonicity. Since the urine sodium concentration is less than that of blood, more water than sodium is lost, causing hypernatremia and hypertonicity. Common iatrogenic solute diuresis occurs in association with mannitol administration, radiocontrast media, and high-protein feedings (enteral or parenteral), leading to increased urea production and excretion. Less commonly, excessive sodium loss may result from cystic renal diseases or Bartter’s syndrome or may develop during a tubulointerstitial process (such as resolving ATN). In these so-called salt-wasting disorders, the tubule damage results in direct impairment of sodium reabsorption and indirectly reduces the responsiveness of the tubule to aldosterone. Usually, the sodium losses are mild, and the obligatory urine output is <2 L/d; resolving ATN and postobstructive diuresis are exceptions and may be associated with significant natriuresis and polyuria.
Formation of large volumes of dilute urine is usually due to polydipsic states or diabetes insipidus. Primary polydipsia can result from habit, psychiatric disorders, neurologic lesions, or medications. During deliberate polydipsia, extracellular fluid volume is normal or expanded and plasma vasopressin levels are reduced because serum osmolality tends to be near the lower limits of normal. Urine osmolality is also maximally dilute at 50 mosmol/L.
Central diabetes insipidus may be idiopathic in origin or secondary to a variety of conditions, including hypophysectomy, trauma, neoplastic, inflammatory, vascular, or infectious hypothalamic diseases. Idiopathic central diabetes insipidus is associated with selective destruction of the vasopressin-secreting neurons in the supraoptic and paraventricular nuclei and can either be inherited as an autosomal dominant trait or occur spontaneously. Nephrogenic diabetes insipidus can occur in a variety of clinical situations, as summarized in Fig. 61-4.
A plasma vasopressin level is recommended as the best method for distinguishing between central and nephrogenic diabetes insipidus. Alternatively, a water deprivation test plus exogenous vasopressin may distinguish primary polydipsia from central and nephrogenic diabetes insipidus. For a detailed discussion, see Chap. 404.
62e | Atlas of Urinary Sediments and Renal Biopsies |
Key diagnostic features of selected diseases in renal biopsy are illustrated, with light, immunofluorescence, and electron microscopic images. Common urinalysis findings are also documented.
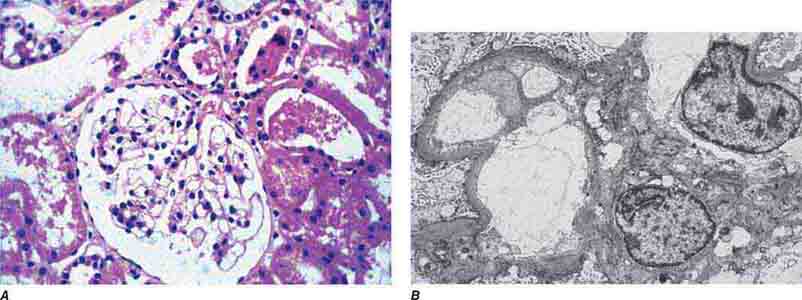
FIGURE 62e-1 Minimal-change disease. In minimal-change disease, light microscopy is unremarkable (A), whereas electron microscopy (B) reveals podocyte injury evidenced by complete foot process effacement. (ABF/Vanderbilt Collection.)
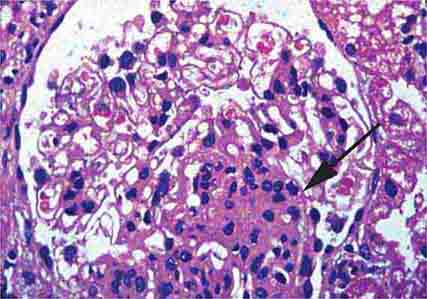
FIGURE 62e-2 Focal segmental glomerulosclerosis (FSGS). There is a well-defined segmental increase in matrix and obliteration of capillary loops (arrow), the sine qua non of segmental sclerosis not otherwise specified (NOS) type. (EGN/UPenn Collection.)
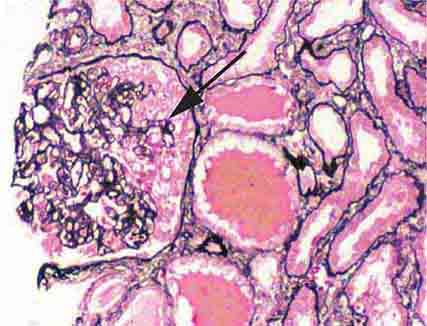
FIGURE 62e-3 Collapsing glomerulopathy. There is segmental collapse (arrow) of the glomerular capillary loops and overlying podocyte hyperplasia. This lesion may be idiopathic or associated with HIV infection and has a particularly poor prognosis. (ABF/Vanderbilt Collection.)
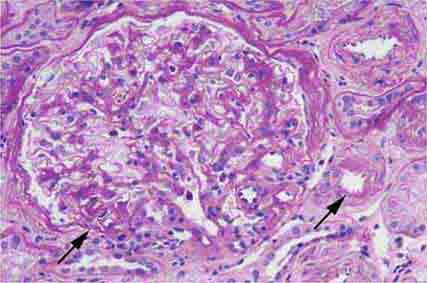
FIGURE 62e-4 Hilar variant of FSGS. There is segmental sclerosis of the glomerular tuft at the vascular pole with associated hyalinosis, also present in the afferent arteriole (arrows). This lesion often occurs as a secondary response when nephron mass is lost due to, e.g., scarring from other conditions. Patients usually have less proteinuria and less steroid response than FSGS, NOS type. (ABF/Vanderbilt Collection.)
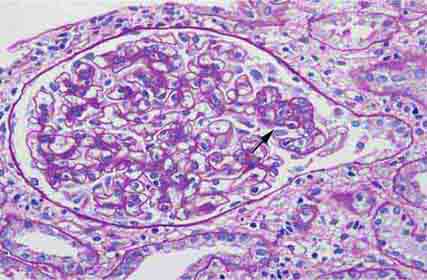
FIGURE 62e-5 Tip lesion variant of FSGS. There is segmental sclerosis of the glomerular capillary loops at the proximal tubular outlet (arrow). This lesion has a better prognosis than other types of FSGS. (ABF/Vanderbilt Collection.)
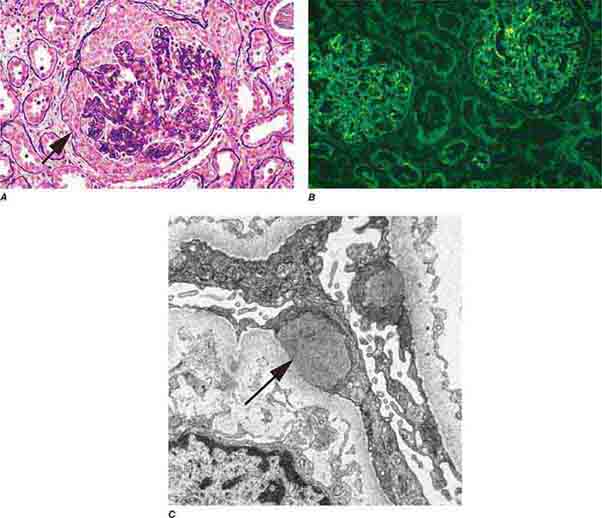
FIGURE 62e-6 Postinfectious (poststreptococcal) glomerulonephritis. The glomerular tuft shows proliferative changes with numerous polymorphonuclear leukocytes (PMNs), with a crescentic reaction (arrow) in severe cases (A). These deposits localize in the mesangium and along the capillary wall in a subepithelial pattern and stain dominantly for C3 and to a lesser extent for IgG (B). Subepithelial hump-shaped deposits are seen by electron microscopy (arrow) (C). (ABF/Vanderbilt Collection.)
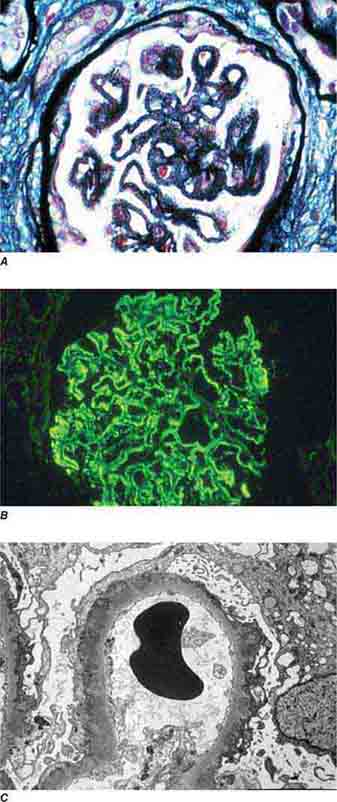
FIGURE 62e-7 Membranous glomerulopathy. Membranous glomerulopathy is due to subepithelial deposits, with resulting basement membrane reaction, resulting in the appearance of spike-like projections on silver stain (A). The deposits are directly visualized by fluorescent anti-IgG, revealing diffuse granular capillary loop staining (B). By electron microscopy, the subepithelial location of the deposits and early surrounding basement membrane reaction is evident, with overlying foot process effacement (C). (ABF/Vanderbilt Collection.)
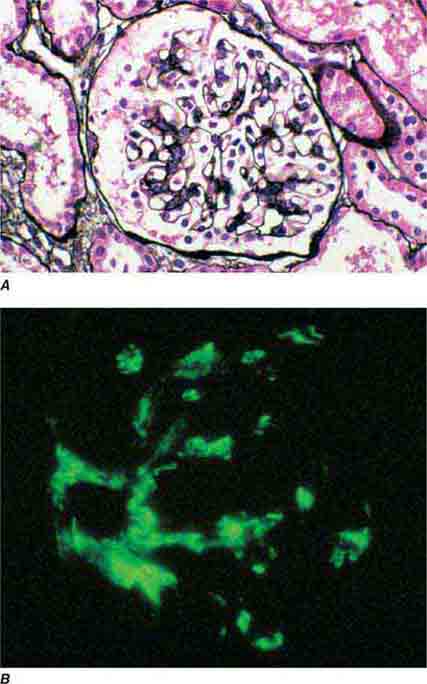
FIGURE 62e-8 IgA nephropathy. There is variable mesangial expansion due to mesangial deposits, with some cases also showing endocapillary proliferation or segmental sclerosis (A). By immunofluorescence, mesangial IgA deposits are evident (B). (ABF/Vanderbilt Collection.)
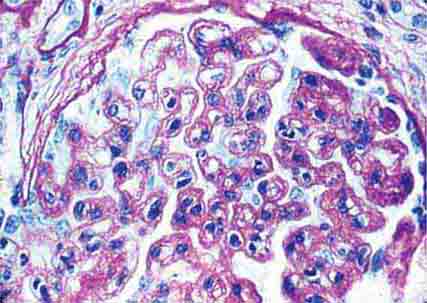
FIGURE 62e-9 Membranoproliferative glomerulonephritis. There is mesangial expansion and endocapillary proliferation with cellular interposition in response to subendothelial deposits, resulting in the “tram-track” of duplication of glomerular basement membrane. (EGN/UPenn Collection.)
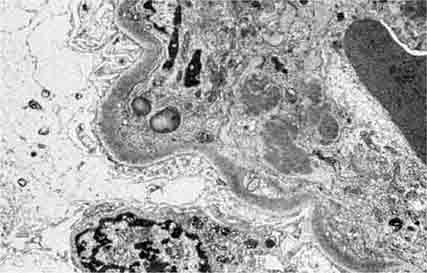
FIGURE 62e-10 Dense deposit disease (membranoproliferative glomerulonephritis type II). By light microscopy, there is a membranoproliferative pattern. By electron microscopy, there is a dense transformation of the glomerular basement membrane with round, globular deposits within the mesangium. By immunofluorescence, only C3 staining is usually present. Dense deposit disease is part of the group of renal diseases called C3 glomerulopathy, related to underlying complement dysregulation. (ABF/Vanderbilt Collection.)
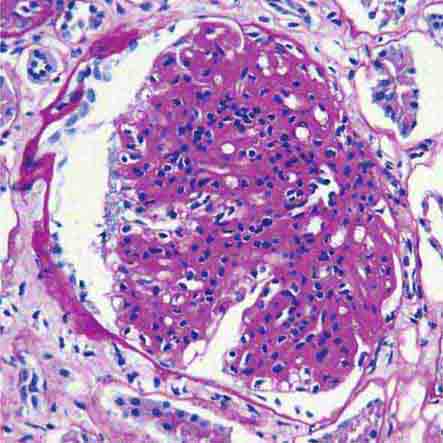
FIGURE 62e-11 C3 glomerulonephritis. By light microscopy, there is a membranoproliferative pattern. C3 glomerulonephritis is part of the group of renal diseases called C3 glomerulopathy, related to underlying complement dysregulation. (ABF/Vanderbilt Collection.)
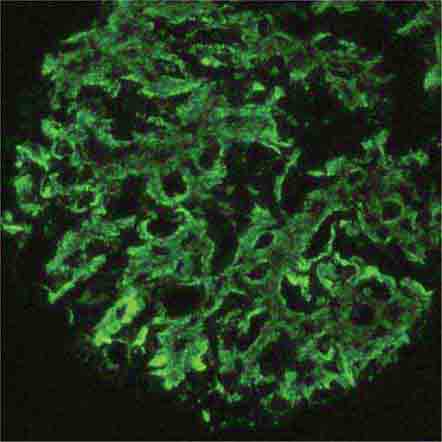
FIGURE 62e-12 C3 glomerulonephritis. By immunofluorescence, only C3 staining is usually present, with occasional minimal immunoglobulin, in an irregular capillary wall and mesangial distribution. (ABF/Vanderbilt Collection.)
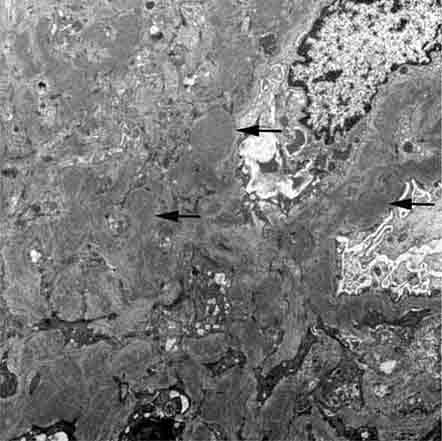
FIGURE 62e-13 C3 glomerulonephritis. By electron microscopy, usual density deposits are present (arrows), including mesangial, subendothelial, and occasional hump-type subepithelial deposits. (ABF/Vanderbilt Collection.)
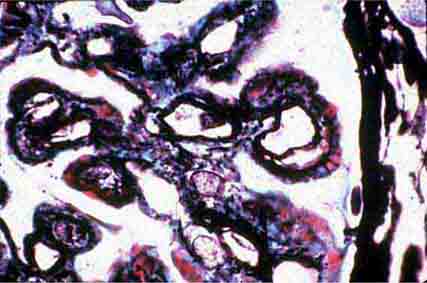
FIGURE 62e-14 Mixed proliferative and membranous glomerulonephritis. This specimen shows pink subepithelial deposits with spike reaction, and the “tram-track” sign of reduplication of glomerular basement membrane, resulting from subendothelial deposits, as may be seen in mixed membranous and proliferative lupus nephritis (International Society of Nephrology [ISN]/Renal Pathology Society [RPS] class V and IV). (EGN/UPenn Collection.)
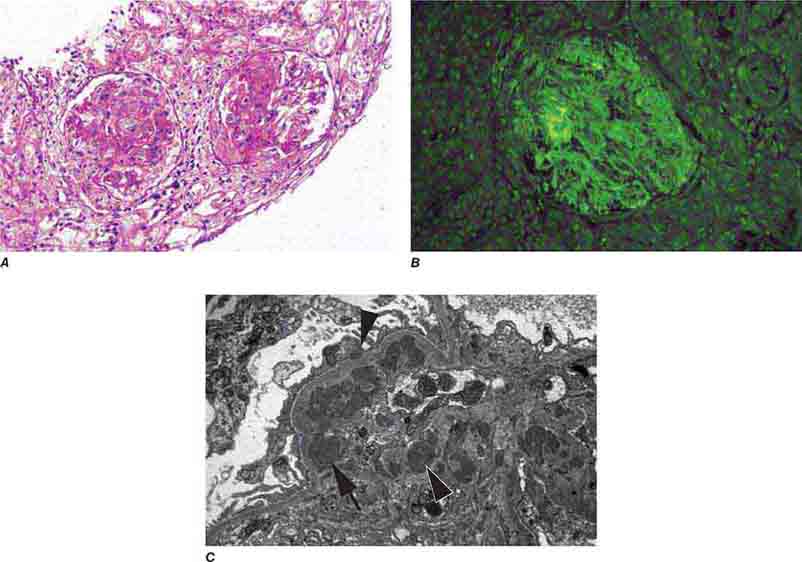
FIGURE 62e-15 Lupus nephritis. Proliferative lupus nephritis, ISN/RPS class III (focal) or IV (diffuse), manifests as endocapillary proliferation, which may result in segmental necrosis due to deposits, particularly in the subendothelial area (A). By immunofluorescence, chunky irregular mesangial and capillary loop deposits are evident, with some of the peripheral loop deposits having a smooth, molded outer contour due to their subendothelial location. These deposits typically stain for all three immunoglobulins, IgG, IgA, IgM, and both C3 and C1q (B). By electron microscopy, subendothelial (arrow), mesangial (white rim arrowhead), and rare subepithelial (black arrowhead) dense immune complex deposits are evident, along with extensive foot process effacement (C). (ABF/Vanderbilt Collection.)
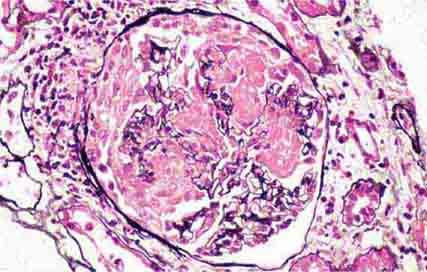
FIGURE 62e-16 Granulomatosis with polyangiitis (Wegener’s). This pauci-immune necrotizing crescentic glomerulonephritis shows numerous breaks in the glomerular basement membrane with associated segmental fibrinoid necrosis and a crescent formed by proliferation of the parietal epithelium. Note that the uninvolved segment of the glomerulus (at ∼5 o’clock) shows no evidence of proliferation or immune complexes. (ABF/Vanderbilt Collection.)
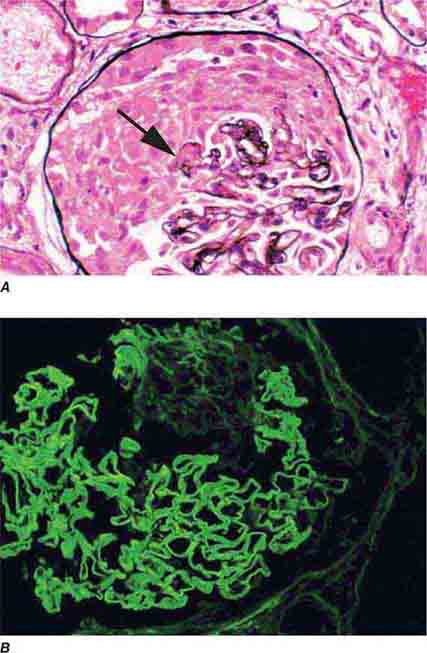
FIGURE 62e-17 Anti–glomerular basement membrane antibody-mediated glomerulonephritis. There is segmental necrosis with a break of the glomerular basement membrane (arrow) and a cellular crescent (A), and immunofluorescence for IgG shows linear staining of the glomerular basement membrane with a small crescent at ∼1 o’clock (B). (ABF/Vanderbilt Collection.)
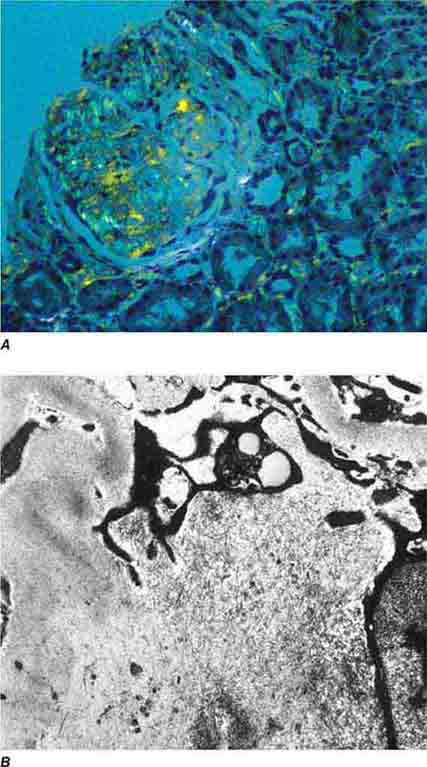
FIGURE 62e-18 Amyloidosis. Amyloidosis shows amorphous, acellular expansion of the mesangium, with material often also infiltrating glomerular basement membranes, vessels, and the interstitium, with apple-green birefringence by polarized Congo red stain (A). The deposits are composed of randomly organized 9- to 11-nm fibrils by electron microscopy (B). (ABF/Vanderbilt Collection.)
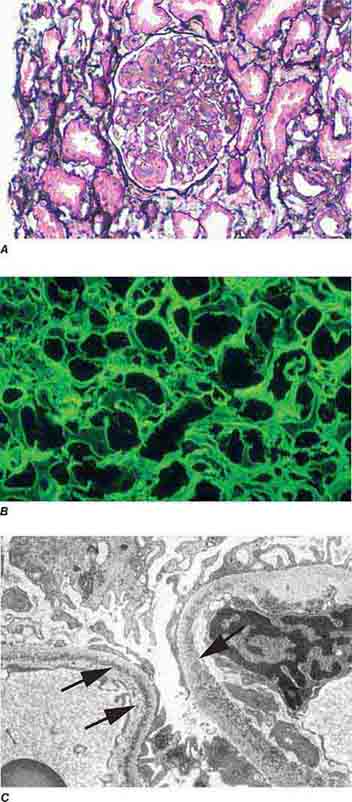
FIGURE 62e-19 Light chain deposition disease. There is mesangial expansion, often nodular by light microscopy (A), with immunofluorescence showing monoclonal staining, more commonly with kappa than lambda light chain, of tubules (B) and glomerular tufts. By electron microscopy (C), the deposits show an amorphous granular appearance and line the inside of the glomerular basement membrane (arrows) and are also found along the tubular basement membranes. (ABF/Vanderbilt Collection.)
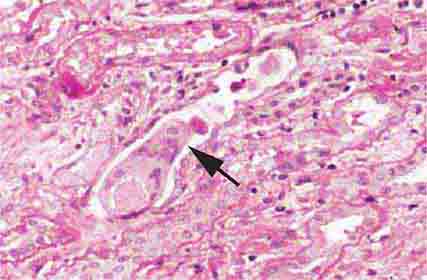
FIGURE 62e-20 Light chain cast nephropathy (myeloma kidney). Monoclonal light chains precipitate in tubules and result in a syncytial giant cell reaction (arrow) surrounding the casts and a surrounding chronic interstitial nephritis with tubulointerstitial fibrosis. (ABF/Vanderbilt Collection.)
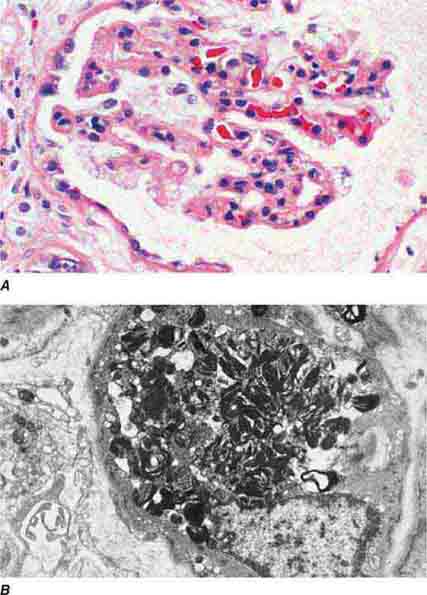
FIGURE 62e-21 Fabry’s disease. Due to deficiency of α-galactosidase, there is abnormal accumulation of glycolipids, resulting in foamy podocytes by light microscopy (A). These deposits can be directly visualized by electron microscopy (B), where the glycosphingolipid appears as whorled so-called myeloid bodies, particularly in the podocytes. (ABF/Vanderbilt Collection.)
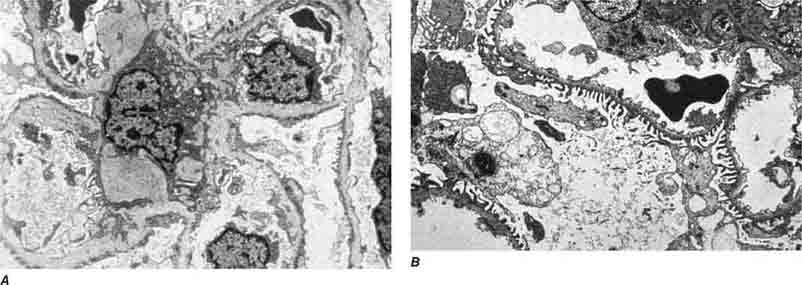
FIGURE 62e-22 Alport’s syndrome and thin glomerular basement membrane lesion. In Alport’s syndrome, there is irregular thinning alternating with thickened so-called basket-weaving abnormal organization of the glomerular basement membrane (A). In benign familial hematuria, or in early cases of Alport’s syndrome or female carriers, only extensive thinning of the glomerular basement membrane is seen by electron microscopy (B). (ABF/Vanderbilt Collection.)
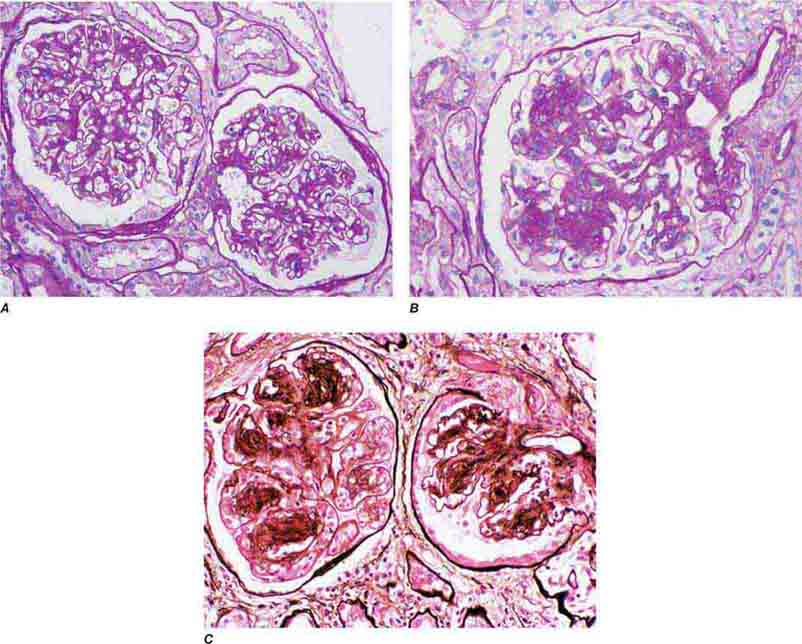
FIGURE 62e-23 Diabetic nephropathy. In the earliest stage of diabetic nephropathy, only mild mesangial increase and prominent glomerular basement membranes (confirmed to be thickened by electron microscopy) are present (A). In slightly more advanced stages, more marked mesangial expansion with early nodule formation develops, with evident arteriolar hyaline (B). In established diabetic nephropathy, there is nodular mesangial expansion, so-called Kimmelstiel-Wilson nodules, with increased mesangial matrix and cellularity, microaneurysm formation in the glomerulus on the left, and prominent glomerular basement membranes without evidence of immune deposits and arteriolar hyalinosis of both afferent and efferent arterioles (C). (ABF/Vanderbilt Collection.)
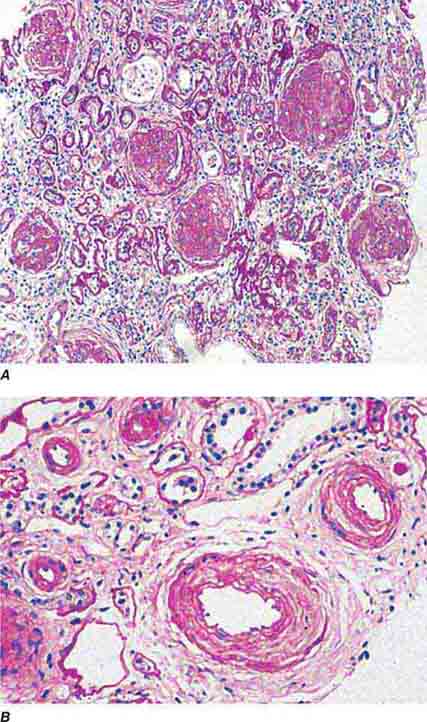
FIGURE 62e-24 Arterionephrosclerosis. Hypertension-associated injury often manifests extensive global sclerosis of glomeruli, with accompanying and proportional tubulointerstitial fibrosis and pericapsular fibrosis, and there may be segmental sclerosis (A). The vessels show disproportionately severe changes of intimal fibrosis, medial hypertrophy, and arteriolar hyaline deposits (B). (ABF/Vanderbilt Collection.)
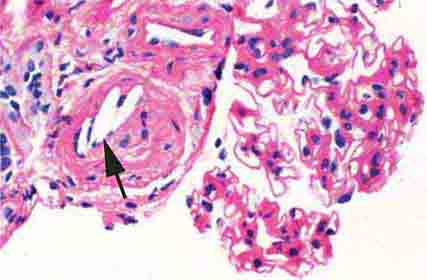
FIGURE 62e-25 Cholesterol emboli. Cholesterol emboli cause cleft-like spaces (arrow) where the lipid has been extracted during processing, with smooth outer contours and surrounding fibrotic and mononuclear cell reaction in these arterioles. (ABF/Vanderbilt Collection.)
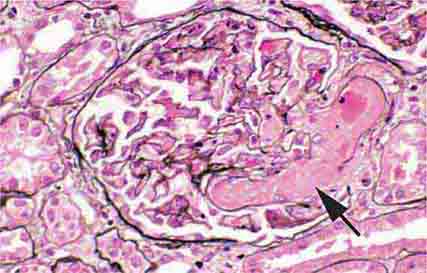
FIGURE 62e-26 Hemolytic-uremic syndrome. There are characteristic intraglomerular fibrin thrombi, with a chunky pink appearance (thrombotic microangiopathy) (arrow). The remaining portion of the capillary tuft shows corrugation of the glomerular basement membrane due to ischemia. (ABF/Vanderbilt Collection.)
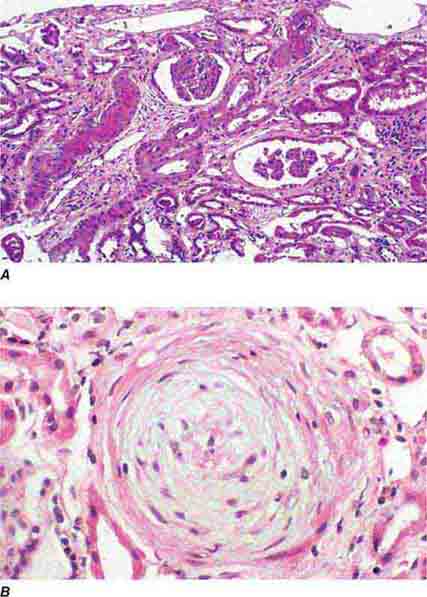
FIGURE 62e-27 Progressive systemic sclerosis. Acutely, there is fibrinoid necrosis of interlobular and larger vessels, with intervening normal vessels and ischemic change in the glomeruli (A). Chronically, this injury leads to intimal proliferation, the so-called onion-skinning appearance (B). (ABF/Vanderbilt Collection.)
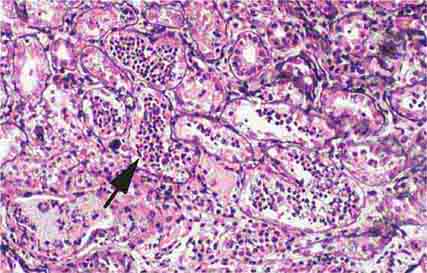
FIGURE 62e-28 Acute pyelonephritis. There are characteristic intratubular plugs and casts of PMNs (arrow) with inflammation extending into the surrounding interstitium and accompanying tubular injury. (ABF/Vanderbilt Collection.)
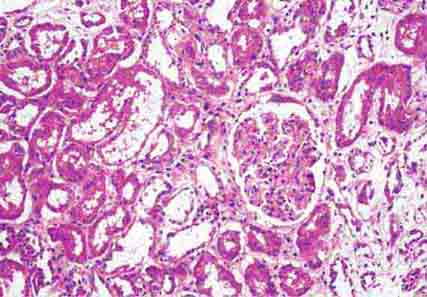
FIGURE 62e-29 Acute tubular injury. There is extensive flattening of the tubular epithelium and loss of the brush border, with mild interstitial edema, characteristic of acute tubular injury due to ischemia. (ABF/Vanderbilt Collection.)
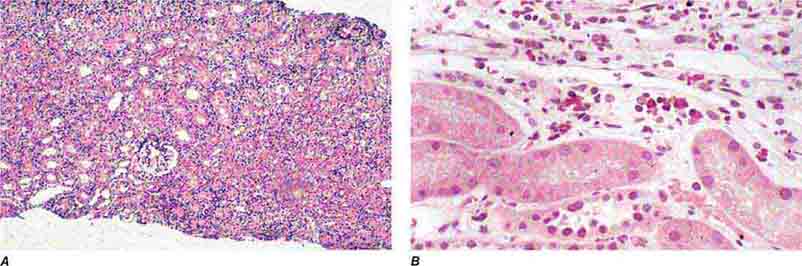
FIGURE 62e-30 Acute interstitial nephritis. There is extensive interstitial lymphoplasmocytic infiltrate with mild edema and associated tubular injury (A), which is frequently associated with interstitial eosinophils (B) when caused by a drug hypersensitivity reaction. (ABF/Vanderbilt Collection.)
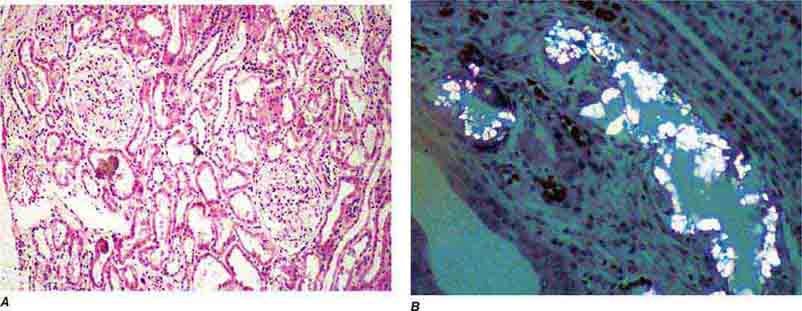
FIGURE 62e-31 Oxalosis. Calcium oxalate crystals have caused extensive tubular injury, with flattening and regeneration of tubular epithelium (A). Crystals are well visualized as sheaves when viewed under polarized light (B). (ABF/Vanderbilt Collection.)
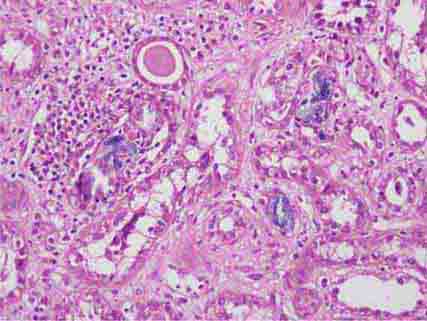
FIGURE 62e-32 Acute phosphate nephropathy. There is extensive acute tubular injury with intratubular nonpolarizable calcium phosphate crystals. (ABF/Vanderbilt Collection.)
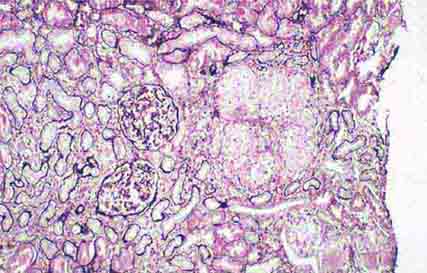
FIGURE 62e-33 Sarcoidosis. There is chronic interstitial nephritis with numerous, confluent, nonnecrotizing granulomas. The glomeruli are unremarkable, but there is moderate tubular atrophy and interstitial fibrosis. (ABF/Vanderbilt Collection.)
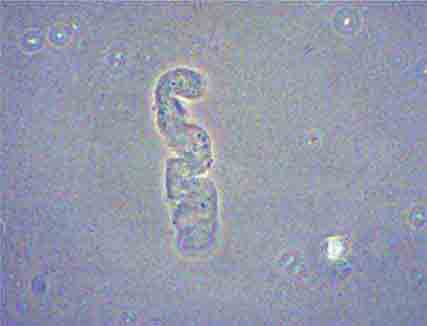
FIGURE 62e-34 Hyaline cast. (ABF/Vanderbilt Collection.)
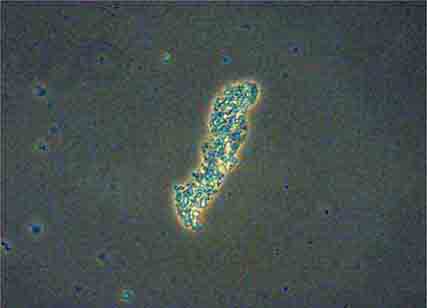
FIGURE 62e-35 Coarse granular cast. (ABF/Vanderbilt Collection.)
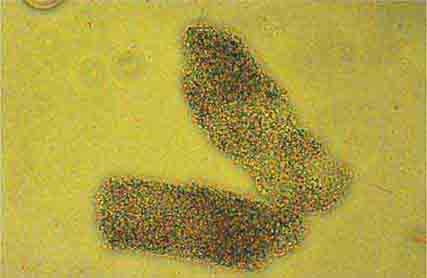
FIGURE 62e-36 Fine granular casts. (ABF/Vanderbilt Collection.)
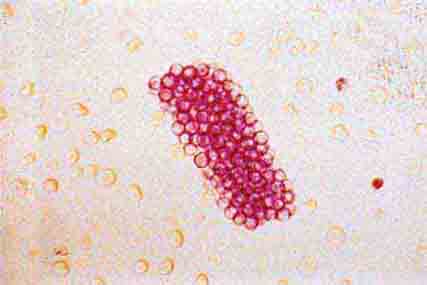
FIGURE 62e-37 Red blood cell cast. (ABF/Vanderbilt Collection.)
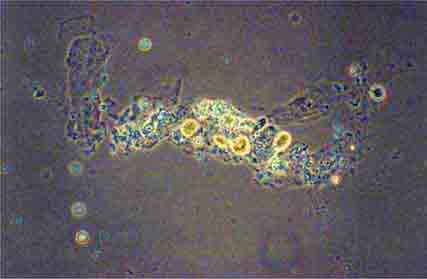
FIGURE 62e-38 White blood cell cast. (ABF/Vanderbilt Collection.)
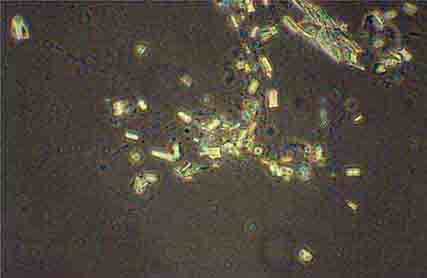
FIGURE 62e-39 Triple phosphate crystals. (ABF/Vanderbilt Collection.)
FIGURE 62e-40 “Maltese cross” formation in an oval fat body. (ABF/Vanderbilt Collection.)

FIGURE 62e-41 Uric acid crystals. (ABF/Vanderbilt Collection.)
63 | Fluid and Electrolyte Disturbances |
SODIUM AND WATER
COMPOSITION OF BODY FLUIDS
Water is the most abundant constituent in the body, comprising approximately 50% of body weight in women and 60% in men. Total-body water is distributed in two major compartments: 55–75% is intracellular (intracellular fluid [ICF]), and 25–45% is extracellular (extracellular fluid [ECF]). The ECF is further subdivided into intravascular (plasma water) and extravascular (interstitial) spaces in a ratio of 1:3. Fluid movement between the intravascular and interstitial spaces occurs across the capillary wall and is determined by Starling forces, i.e., capillary hydraulic pressure and colloid osmotic pressure. The transcapillary hydraulic pressure gradient exceeds the corresponding oncotic pressure gradient, thereby favoring the movement of plasma ultrafiltrate into the extravascular space. The return of fluid into the intravascular compartment occurs via lymphatic flow.
The solute or particle concentration of a fluid is known as its osmolality, expressed as milliosmoles per kilogram of water (mOsm/kg). Water easily diffuses across most cell membranes to achieve osmotic equilibrium (ECF osmolality = ICF osmolality). Notably, the extracellular and intracellular solute compositions differ considerably owing to the activity of various transporters, channels, and ATP-driven membrane pumps. The major ECF particles are Na+ and its accompanying anions Cl– and HCO3–, whereas K+ and organic phosphate esters (ATP, creatine phosphate, and phospholipids) are the predominant ICF osmoles. Solutes that are restricted to the ECF or the ICF determine the “tonicity” or effective osmolality of that compartment. Certain solutes, particularly urea, do not contribute to water shifts across most membranes and are thus known as ineffective osmoles.
Water Balance Vasopressin secretion, water ingestion, and renal water transport collaborate to maintain human body fluid osmolality between 280 and 295 mOsm/kg. Vasopressin (AVP) is synthesized in magnocellular neurons within the hypothalamus; the distal axons of these neurons project to the posterior pituitary or neurohypophysis, from which AVP is released into the circulation. A network of central “osmoreceptor” neurons, which includes the AVP-expressing magnocellular neurons themselves, sense circulating osmolality via nonselective, stretch-activated cation channels. These osmoreceptor neurons are activated or inhibited by modest increases and decreases in circulating osmolality, respectively; activation leads to AVP release and thirst.
AVP secretion is stimulated as systemic osmolality increases above a threshold level of ~285 mOsm/kg, above which there is a linear relationship between osmolality and circulating AVP (Fig. 63-1). Thirst and thus water ingestion are also activated at ~285 mOsm/kg, beyond which there is an equivalent linear increase in the perceived intensity of thirst as a function of circulating osmolality. Changes in blood volume and blood pressure are also direct stimuli for AVP release and thirst, albeit with a less sensitive response profile. Of perhaps greater clinical relevance to the pathophysiology of water homeostasis, ECF volume strongly modulates the relationship between circulating osmolality and AVP release, such that hypovolemia reduces the osmotic threshold and increases the slope of the response curve to osmolality; hypervolemia has an opposite effect, increasing the osmotic threshold and reducing the slope of the response curve (Fig. 63-1). Notably, AVP has a half-life in the circulation of only 10–20 minutes; thus, changes in ECF volume and/or circulating osmolality can rapidly affect water homeostasis. In addition to volume status, a number of other “nonosmotic” stimuli have potent activating effects on osmosensitive neurons and AVP release, including nausea, intracerebral angiotensin II, serotonin, and multiple drugs.
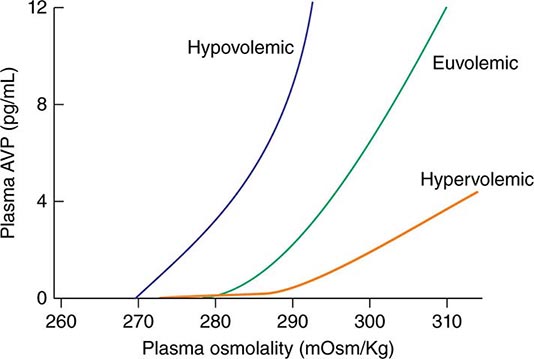
FIGURE 63-1 Circulating levels of vasopressin (AVP) in response to changes in osmolality. Plasma AVP becomes detectable in euvolemic, healthy individuals at a threshold of ~285 mOsm/kg, above which there is a linear relationship between osmolality and circulating AVP. The vasopressin response to osmolality is modulated strongly by volume status. The osmotic threshold is thus slightly lower in hypovolemia, with a steeper response curve; hypervolemia reduces the sensitivity of circulating AVP levels to osmolality.
The excretion or retention of electrolyte-free water by the kidney is modulated by circulating AVP. AVP acts on renal, V2-type receptors in the thick ascending limb of Henle and principal cells of the collecting duct (CD), increasing intracellular levels of cyclic AMP and activating protein kinase A (PKA)–dependent phosphorylation of multiple transport proteins. The AVP- and PKA-dependent activation of Na+-Cl– and K+ transport by the thick ascending limb of the loop of Henle (TALH) is a key participant in the countercurrent mechanism (Fig. 63-2). The countercurrent mechanism ultimately increases the interstitial osmolality in the inner medulla of the kidney, driving water absorption across the renal CD. However, water, salt, and solute transport by both proximal and distal nephron segments participates in the renal concentrating mechanism (Fig. 63-2). Water transport across apical and basolateral aquaporin-1 water channels in the descending thin limb of the loop of Henle is thus involved, as is passive absorption of Na+-Cl– by the thin ascending limb, via apical and basolateral CLC-K1 chloride channels and paracellular Na+ transport. Renal urea transport in turn plays important roles in the generation of the medullary osmotic gradient and the ability to excrete solute-free water under conditions of both high and low protein intake (Fig. 63-2).
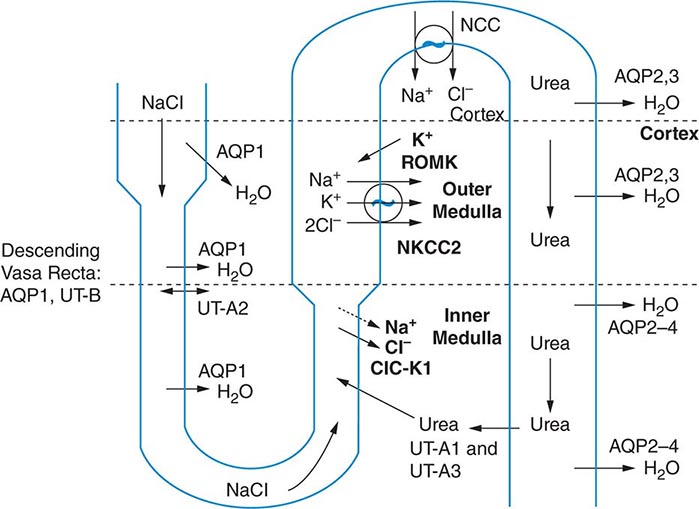
FIGURE 63-2 The renal concentrating mechanism. Water, salt, and solute transport by both proximal and distal nephron segments participates in the renal concentrating mechanism (see text for details). Diagram showing the location of the major transport proteins involved; a loop of Henle is depicted on the left, collecting duct on the right. AQP, aquaporin; CLC-K1, chloride channel; NKCC2, Na-K-2Cl cotransporter; ROMK, renal outer medullary K+ channel; UT, urea transporter. (Used with permission from JM Sands: Molecular approaches to urea transporters. J Am Soc Nephrol 13:2795, 2002.)
AVP-induced, PKA-dependent phosphorylation of the aquaporin-2 water channel in principal cells stimulates the insertion of active water channels into the lumen of the CD, resulting in transepithelial water absorption down the medullary osmotic gradient (Fig. 63-3). Under “antidiuretic” conditions, with increased circulating AVP, the kidney reabsorbs water filtered by the glomerulus, equilibrating the osmolality across the CD epithelium to excrete a hypertonic, “concentrated” urine (osmolality of up to 1200 mOsm/kg). In the absence of circulating AVP, insertion of aquaporin-2 channels and water absorption across the CD is essentially abolished, resulting in secretion of a hypotonic, dilute urine (osmolality as low as 30–50 mOsm/kg). Abnormalities in this “final common pathway” are involved in most disorders of water homeostasis, e.g., a reduced or absent insertion of active aquaporin-2 water channels into the membrane of principal cells in diabetes insipidus.
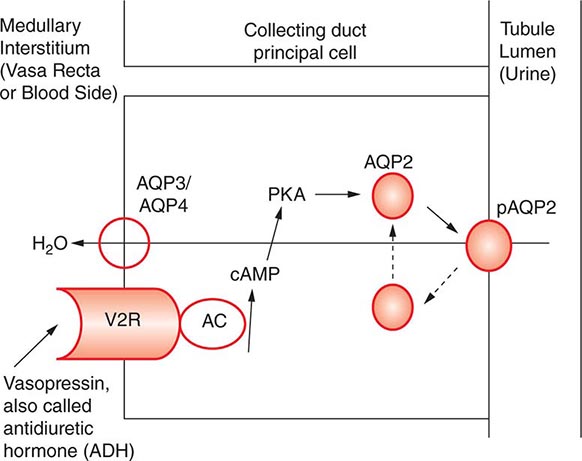
FIGURE 63-3 Vasopressin and the regulation of water permeability in the renal collecting duct. Vasopressin binds to the type 2 vasopressin receptor (V2R) on the basolateral membrane of principal cells, activates adenylyl cyclase (AC), increases intracellular cyclic adenosine monophosphatase (cAMP), and stimulates protein kinase A (PKA) activity. Cytoplasmic vesicles carrying aquaporin-2 (AQP) water channel proteins are inserted into the luminal membrane in response to vasopressin, thereby increasing the water permeability of this membrane. When vasopressin stimulation ends, water channels are retrieved by an endocytic process and water permeability returns to its low basal rate. The AQP3 and AQP4 water channels are expressed on the basolateral membrane and complete the transcellular pathway for water reabsorption. pAQP2, phosphorylated aquaporin-2. (From JM Sands, DG Bichet: Nephrogenic diabetes insipidus. Ann Intern Med 144:186, 2006, with permission.)
Maintenance of Arterial Circulatory Integrity Sodium is actively pumped out of cells by the Na+/K+-ATPase membrane pump. In consequence, 85–90% of body Na+ is extracellular, and the ECF volume (ECFV) is a function of total-body Na+ content. Arterial perfusion and circulatory integrity are, in turn, determined by renal Na+ retention or excretion, in addition to the modulation of systemic arterial resistance. Within the kidney, Na+ is filtered by the glomeruli and then sequentially reabsorbed by the renal tubules. The Na+ cation is typically reabsorbed with the chloride anion (Cl–), and, thus, chloride homeostasis also affects the ECFV. On a quantitative level, at a glomerular filtration rate (GFR) of 180 L/d and serum Na+ of ~140 mM, the kidney filters some 25,200 mmol/d of Na+. This is equivalent to ~1.5 kg of salt, which would occupy roughly 10 times the extracellular space; 99.6% of filtered Na+-Cl– must be reabsorbed to excrete 100 mM per day. Minute changes in renal Na+-Cl– excretion will thus have significant effects on the ECFV, leading to edema syndromes or hypovolemia.
Approximately two-thirds of filtered Na+-Cl– is reabsorbed by the renal proximal tubule, via both paracellular and transcellular mechanisms. The TALH subsequently reabsorbs another 25–30% of filtered Na+-Cl– via the apical, furosemide-sensitive Na+-K+-2Cl– cotransporter. The adjacent aldosterone-sensitive distal nephron, comprising the distal convoluted tubule (DCT), connecting tubule (CNT), and CD, accomplishes the “fine-tuning” of renal Na+-Cl– excretion. The thiazide-sensitive apical Na+-Cl– cotransporter (NCC) reabsorbs 5–10% of filtered Na+-Cl– in the DCT. Principal cells in the CNT and CD reabsorb Na+ via electrogenic, amiloride-sensitive epithelial Na+ channels (ENaC); Cl– ions are primarily reabsorbed by adjacent intercalated cells, via apical Cl– exchange (Cl–-OH– and Cl–-HCO3– exchange, mediated by the SLC26A4 anion exchanger) (Fig. 63-4).
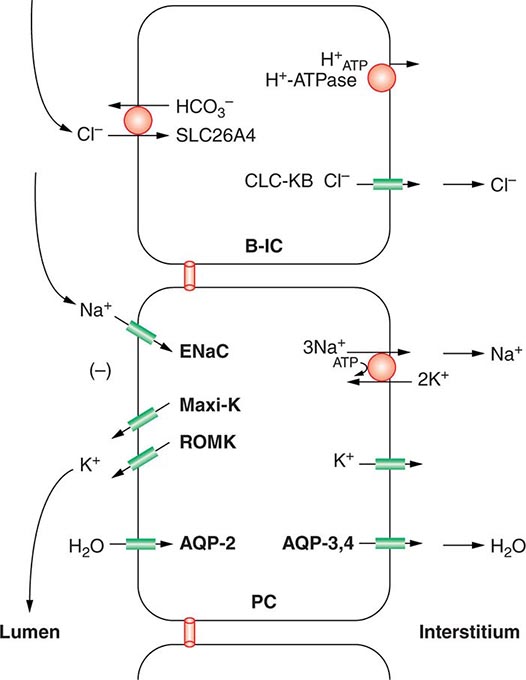
FIGURE 63-4 Sodium, water, and potassium transport in principal cells (PC) and adjacent β-intercalated cells (B-IC). The absorption of Na+ via the amiloride-sensitive epithelial sodium channel (ENaC) generates a lumen-negative potential difference, which drives K+ excretion through the apical secretory K+ channel ROMK (renal outer medullary K+ channel) and/or the flow-dependent BK channel. Transepithelial Cl– transport occurs in adjacent β-intercalated cells, via apical Cl–-HCO3– and Cl–-OH– exchange (SLC26A4 anion exchanger, also known as pendrin) basolateral CLC chloride channels. Water is absorbed down the osmotic gradient by principal cells, through the apical aquaporin-2 (AQP-2) and basolateral aquaporin-3 and aquaporin-4 (Fig. 63-3).
Renal tubular reabsorption of filtered Na+-Cl– is regulated by multiple circulating and paracrine hormones, in addition to the activity of renal nerves. Angiotensin II activates proximal Na+-Cl– reabsorption, as do adrenergic receptors under the influence of renal sympathetic innervation; locally generated dopamine, in contrast, has a natriuretic effect. Aldosterone primarily activates Na+-Cl– reabsorption within the aldosterone-sensitive distal nephron. In particular, aldosterone activates the ENaC channel in principal cells, inducing Na+ absorption and promoting K+ excretion (Fig. 63-4).
Circulatory integrity is critical for the perfusion and function of vital organs. “Underfilling” of the arterial circulation is sensed by ventricular and vascular pressure receptors, resulting in a neurohumoral activation (increased sympathetic tone, activation of the renin-angiotensin-aldosterone axis, and increased circulating AVP) that synergistically increases renal Na+-Cl– reabsorption, vascular resistance, and renal water reabsorption. This occurs in the context of decreased cardiac output, as occurs in hypovolemic states, low-output cardiac failure, decreased oncotic pressure, and/or increased capillary permeability. Alternatively, excessive arterial vasodilation results in relative arterial underfilling, leading to neurohumoral activation in the defense of tissue perfusion. These physiologic responses play important roles in many of the disorders discussed in this chapter. In particular, it is important to appreciate that AVP functions in the defense of circulatory integrity, inducing vasoconstriction, increasing sympathetic nervous system tone, increasing renal retention of both water and Na+-Cl–, and modulating the arterial baroreceptor reflex. Most of these responses involve activation of systemic V1A AVP receptors, but concomitant activation of V2 receptors in the kidney can result in renal water retention and hyponatremia.
HYPOVOLEMIA
Etiology True volume depletion, or hypovolemia, generally refers to a state of combined salt and water loss, leading to contraction of the ECFV. The loss of salt and water may be renal or nonrenal in origin.
RENAL CAUSES Excessive urinary Na+-Cl– and water loss is a feature of several conditions. A high filtered load of endogenous solutes, such as glucose and urea, can impair tubular reabsorption of Na+-Cl– and water, leading to an osmotic diuresis. Exogenous mannitol, often used to decrease intracerebral pressure, is filtered by glomeruli but not reabsorbed by the proximal tubule, thus causing an osmotic diuresis. Pharmacologic diuretics selectively impair Na+-Cl– reabsorption at specific sites along the nephron, leading to increased urinary Na+-Cl– excretion. Other drugs can induce natriuresis as a side effect. For example, acetazolamide can inhibit proximal tubular Na+-Cl– absorption via its inhibition of carbonic anhydrase; other drugs, such as the antibiotics trimethoprim and pentamidine, inhibit distal tubular Na+ reabsorption through the amiloride-sensitive ENaC channel, leading to urinary Na+-Cl– loss. Hereditary defects in renal transport proteins are also associated with reduced reabsorption of filtered Na+-Cl– and/or water. Alternatively, mineralocorticoid deficiency, mineralocorticoid resistance, or inhibition of the mineralocorticoid receptor (MLR) can reduce Na+-Cl– reabsorption by the aldosterone-sensitive distal nephron. Finally, tubulointerstitial injury, as occurs in interstitial nephritis, acute tubular injury, or obstructive uropathy, can reduce distal tubular Na+-Cl– and/or water absorption.
Excessive excretion of free water, i.e., water without electrolytes, can also lead to hypovolemia. However, the effect on ECFV is usually less marked, given that two-thirds of the water volume is lost from the ICF. Excessive renal water excretion occurs in the setting of decreased circulating AVP or renal resistance to AVP (central and nephrogenic diabetes insipidus, respectively).
EXTRARENAL CAUSES Nonrenal causes of hypovolemia include fluid loss from the gastrointestinal tract, skin, and respiratory system. Accumulations of fluid within specific tissue compartments, typically the interstitium, peritoneum, or gastrointestinal tract, can also cause hypovolemia.
Approximately 9 L of fluid enter the gastrointestinal tract daily, 2 L by ingestion and 7 L by secretion; almost 98% of this volume is absorbed, such that daily fecal fluid loss is only 100–200 mL. Impaired gastrointestinal reabsorption or enhanced secretion of fluid can cause hypovolemia. Because gastric secretions have a low pH (high H+ concentration), whereas biliary, pancreatic, and intestinal secretions are alkaline (high HCO3– concentration), vomiting and diarrhea are often accompanied by metabolic alkalosis and acidosis, respectively.
Evaporation of water from the skin and respiratory tract (so-called “insensible losses”) constitutes the major route for loss of solute-free water, which is typically 500–650 mL/d in healthy adults. This evaporative loss can increase during febrile illness or prolonged heat exposure. Hyperventilation can also increase insensible losses via the respiratory tract, particularly in ventilated patients; the humidity of inspired air is another determining factor. In addition, increased exertion and/or ambient temperature will increase insensible losses via sweat, which is hypotonic to plasma. Profuse sweating without adequate repletion of water and Na+-Cl– can thus lead to both hypovolemia and hypertonicity. Alternatively, replacement of these insensible losses with a surfeit of free water, without adequate replacement of electrolytes, may lead to hypovolemic hyponatremia.
Excessive fluid accumulation in interstitial and/or peritoneal spaces can also cause intravascular hypovolemia. Increases in vascular permeability and/or a reduction in oncotic pressure (hypoalbuminemia) alter Starling forces, resulting in excessive “third spacing” of the ECFV. This occurs in sepsis syndrome, burns, pancreatitis, nutritional hypoalbuminemia, and peritonitis. Alternatively, distributive hypovolemia can occur due to accumulation of fluid within specific compartments, for example within the bowel lumen in gastrointestinal obstruction or ileus. Hypovolemia can also occur after extracorporeal hemorrhage or after significant hemorrhage into an expandable space, for example, the retroperitoneum.
Diagnostic Evaluation A careful history will usually determine the etiologic cause of hypovolemia. Symptoms of hypovolemia are nonspecific and include fatigue, weakness, thirst, and postural dizziness; more severe symptoms and signs include oliguria, cyanosis, abdominal and chest pain, and confusion or obtundation. Associated electrolyte disorders may cause additional symptoms, for example, muscle weakness in patients with hypokalemia. On examination, diminished skin turgor and dry oral mucous membranes are less than ideal markers of a decreased ECFV in adult patients; more reliable signs of hypovolemia include a decreased jugular venous pressure (JVP), orthostatic tachycardia (an increase of >15–20 beats/min upon standing), and orthostatic hypotension (a >10–20 mmHg drop in blood pressure on standing). More severe fluid loss leads to hypovolemic shock, with hypotension, tachycardia, peripheral vasoconstriction, and peripheral hypoperfusion; these patients may exhibit peripheral cyanosis, cold extremities, oliguria, and altered mental status.
Routine chemistries may reveal an increase in blood urea nitrogen (BUN) and creatinine, reflective of a decrease in GFR. Creatinine is the more dependable measure of GFR, because BUN levels may be influenced by an increase in tubular reabsorption (“prerenal azotemia”), an increase in urea generation in catabolic states, hyperalimentation, or gastrointestinal bleeding, and/or a decreased urea generation in decreased protein intake. In hypovolemic shock, liver function tests and cardiac biomarkers may show evidence of hepatic and cardiac ischemia, respectively. Routine chemistries and/or blood gases may reveal evidence of acid-base disorders. For example, bicarbonate loss due to diarrheal illness is a very common cause of metabolic acidosis; alternatively, patients with severe hypovolemic shock may develop lactic acidosis with an elevated anion gap.
The neurohumoral response to hypovolemia stimulates an increase in renal tubular Na+ and water reabsorption. Therefore, the urine Na+ concentration is typically <20 mM in nonrenal causes of hypovolemia, with a urine osmolality of >450 mOsm/kg. The reduction in both GFR and distal tubular Na+ delivery may cause a defect in renal potassium excretion, with an increase in plasma K+ concentration. Of note, patients with hypovolemia and a hypochloremic alkalosis due to vomiting, diarrhea, or diuretics will typically have a urine Na+ concentration >20 mM and urine pH of >7.0, due to the increase in filtered HCO3–; the urine Cl– concentration in this setting is a more accurate indicator of volume status, with a level <25 mM suggestive of hypovolemia. The urine Na+ concentration is often >20 mM in patients with renal causes of hypovolemia, such as acute tubular necrosis; similarly, patients with diabetes insipidus will have an inappropriately dilute urine.
SODIUM DISORDERS
Disorders of serum Na+ concentration are caused by abnormalities in water homeostasis, leading to changes in the relative ratio of Na+ to body water. Water intake and circulating AVP constitute the two key effectors in the defense of serum osmolality; defects in one or both of these two defense mechanisms cause most cases of hyponatremia and hypernatremia. In contrast, abnormalities in sodium homeostasis per se lead to a deficit or surplus of whole-body Na+-Cl– content, a key determinant of the ECFV and circulatory integrity. Notably, volume status also modulates the release of AVP by the posterior pituitary, such that hypovolemia is associated with higher circulating levels of the hormone at each level of serum osmolality. Similarly, in “hypervolemic” causes of arterial underfilling, e.g., heart failure and cirrhosis, the associated neurohumoral activation is associated with an increase in circulating AVP, leading to water retention and hyponatremia. Therefore, a key concept in sodium disorders is that the absolute plasma Na+ concentration tells one nothing about the volume status of a given patient, which furthermore must be taken into account in the diagnostic and therapeutic approach.
HYPONATREMIA
Hyponatremia, which is defined as a plasma Na+ concentration <135 mM, is a very common disorder, occurring in up to 22% of hospitalized patients. This disorder is almost always the result of an increase in circulating AVP and/or increased renal sensitivity to AVP, combined with an intake of free water; a notable exception is hyponatremia due to low solute intake (see below). The underlying pathophysiology for the exaggerated or “inappropriate” AVP response differs in patients with hyponatremia as a function of their ECFV. Hyponatremia is thus subdivided diagnostically into three groups, depending on clinical history and volume status, i.e., “hypovolemic,” “euvolemic,” and “hypervolemic” (Fig. 63-5).
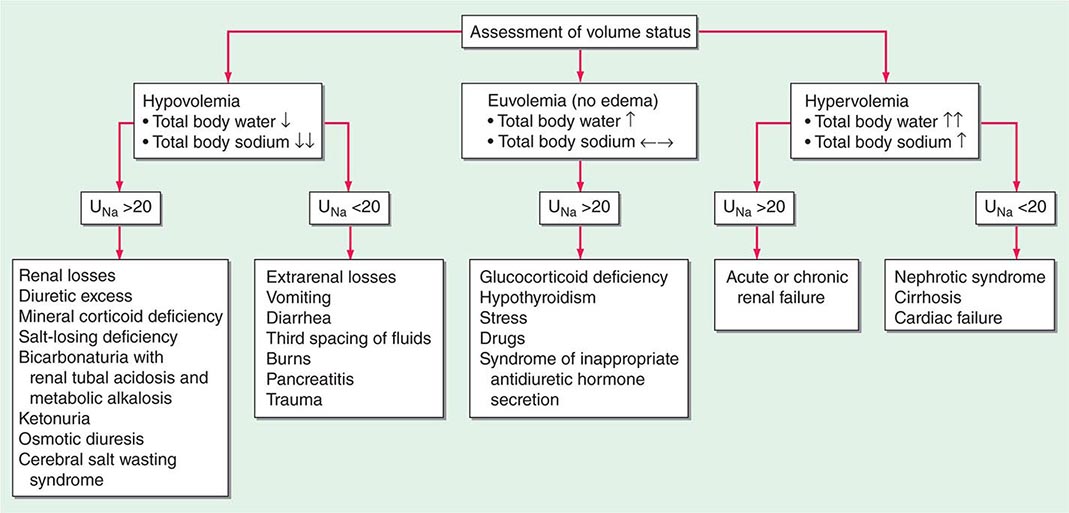
FIGURE 63-5 The diagnostic approach to hyponatremia. (From S Kumar, T Berl: Diseases of water metabolism, in Atlas of Diseases of the Kidney, RW Schrier [ed]. Philadelphia, Current Medicine, Inc, 1999; with permission.)
Hypovolemic Hyponatremia Hypovolemia causes a marked neurohumoral activation, increasing circulating levels of AVP. The increase in circulating AVP helps preserve blood pressure via vascular and baroreceptor V1A receptors and increases water reabsorption via renal V2 receptors; activation of V2 receptors can lead to hyponatremia in the setting of increased free water intake. Nonrenal causes of hypovolemic hyponatremia include GI loss (e.g., vomiting, diarrhea, tube drainage) and insensible loss (sweating, burns) of Na+-Cl– and water, in the absence of adequate oral replacement; urine Na+ concentration is typically <20 mM. Notably, these patients may be clinically classified as euvolemic, with only the reduced urinary Na+ concentration to indicate the cause of their hyponatremia. Indeed, a urine Na+ concentration <20 mM, in the absence of a cause of hypervolemic hyponatremia, predicts a rapid increase in plasma Na+ concentration in response to intravenous normal saline; saline therapy thus induces a water diuresis in this setting, as circulating AVP levels plummet.
The renal causes of hypovolemic hyponatremia share an inappropriate loss of Na+-Cl– in the urine, leading to volume depletion and an increase in circulating AVP; urine Na+ concentration is typically >20 mM (Fig. 63-5). A deficiency in circulating aldosterone and/or its renal effects can lead to hyponatremia in primary adrenal insufficiency and other causes of hypoaldosteronism; hyperkalemia and hyponatremia in a hypotensive and/or hypovolemic patient with high urine Na+ concentration (much greater than 20 mM) should strongly suggest this diagnosis. Salt-losing nephropathies may lead to hyponatremia when sodium intake is reduced, due to impaired renal tubular function; typical causes include reflux nephropathy, interstitial nephropathies, postobstructive uropathy, medullary cystic disease, and the recovery phase of acute tubular necrosis. Thiazide diuretics cause hyponatremia via a number of mechanisms, including polydipsia and diuretic-induced volume depletion. Notably, thiazides do not inhibit the renal concentrating mechanism, such that circulating AVP retains a full effect on renal water retention. In contrast, loop diuretics, which are less frequently associated with hyponatremia, inhibit Na+-Cl– and K+ absorption by the TALH, blunting the countercurrent mechanism and reducing the ability to concentrate the urine. Increased excretion of an osmotically active nonreabsorbable or poorly reabsorbable solute can also lead to volume depletion and hyponatremia; important causes include glycosuria, ketonuria (e.g., in starvation or in diabetic or alcoholic ketoacidosis), and bicarbonaturia (e.g., in renal tubular acidosis or metabolic alkalosis, where the associated bicarbonaturia leads to loss of Na+).
Finally, the syndrome of “cerebral salt wasting” is a rare cause of hypovolemic hyponatremia, encompassing hyponatremia with clinical hypovolemia and inappropriate natriuresis in association with intracranial disease; associated disorders include subarachnoid hemorrhage, traumatic brain injury, craniotomy, encephalitis, and meningitis. Distinction from the more common syndrome of inappropriate antidiuresis is critical because cerebral salt wasting will typically respond to aggressive Na+-Cl– repletion.
Hypervolemic Hyponatremia Patients with hypervolemic hyponatremia develop an increase in total-body Na+-Cl– that is accompanied by a proportionately greater increase in total-body water, leading to a reduced plasma Na+ concentration. As in hypovolemic hyponatremia, the causative disorders can be separated by the effect on urine Na+ concentration, with acute or chronic renal failure uniquely associated with an increase in urine Na+ concentration (Fig. 63-5). The pathophysiology of hyponatremia in the sodium-avid edematous disorders (congestive heart failure [CHF], cirrhosis, and nephrotic syndrome) is similar to that in hypovolemic hyponatremia, except that arterial filling and circulatory integrity is decreased due to the specific etiologic factors (e.g., cardiac dysfunction in CHF, peripheral vasodilation in cirrhosis). Urine Na+ concentration is typically very low, i.e., <10 mM, even after hydration with normal saline; this Na+-avid state may be obscured by diuretic therapy. The degree of hyponatremia provides an indirect index of the associated neurohumoral activation and is an important prognostic indicator in hypervolemic hyponatremia.
Euvolemic Hyponatremia Euvolemic hyponatremia can occur in moderate to severe hypothyroidism, with correction after achieving a euthyroid state. Severe hyponatremia can also be a consequence of secondary adrenal insufficiency due to pituitary disease; whereas the deficit in circulating aldosterone in primary adrenal insufficiency causes hypovolemic hyponatremia, the predominant glucocorticoid deficiency in secondary adrenal failure is associated with euvolemic hyponatremia. Glucocorticoids exert a negative feedback on AVP release by the posterior pituitary such that hydrocortisone replacement in these patients will rapidly normalize the AVP response to osmolality, reducing circulating AVP.
The syndrome of inappropriate antidiuresis (SIAD) is the most frequent cause of euvolemic hyponatremia (Table 63-1). The generation of hyponatremia in SIAD requires an intake of free water, with persistent intake at serum osmolalities that are lower than the usual threshold for thirst; as one would expect, the osmotic threshold and osmotic response curves for the sensation of thirst are shifted downward in patients with SIAD. Four distinct patterns of AVP secretion have been recognized in patients with SIAD, independent for the most part of the underlying cause. Unregulated, erratic AVP secretion is seen in about a third of patients, with no obvious correlation between serum osmolality and circulating AVP levels. Other patients fail to suppress AVP secretion at lower serum osmolalities, with a normal response curve to hyperosmolar conditions; others have a “reset osmostat,” with a lower threshold osmolality and a left-shifted osmotic response curve. Finally, the fourth subset of patients have essentially no detectable circulating AVP, suggesting either a gain in function in renal water reabsorption or a circulating antidiuretic substance that is distinct from AVP. Gain-in-function mutations of a single specific residue in the V2 AVP receptor have been described in some of these patients, leading to constitutive activation of the receptor in the absence of AVP and “nephrogenic” SIAD.
CAUSES OF THE SYNDROME OF INAPPROPRIATE ANTIDIURESIS (SIAD) |
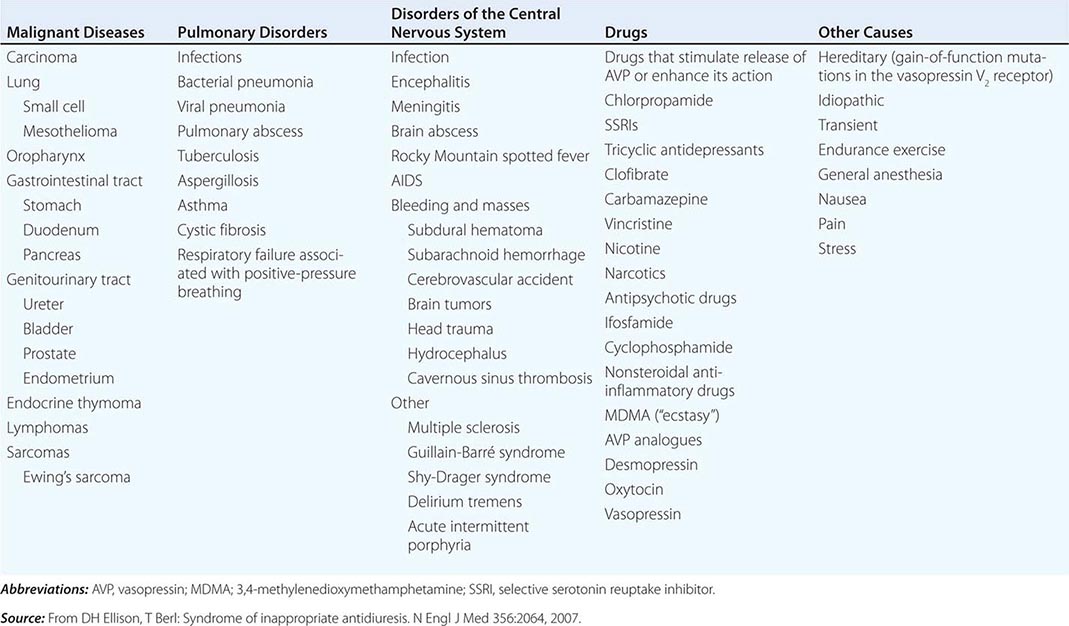
Strictly speaking, patients with SIAD are not euvolemic but are subclinically volume-expanded, due to AVP-induced water and Na+-Cl– retention; “AVP escape” mechanisms invoked by sustained increases in AVP serve to limit distal renal tubular transport, preserving a modestly hypervolemic steady state. Serum uric acid is often low (<4 mg/dL) in patients with SIAD, consistent with suppressed proximal tubular transport in the setting of increased distal tubular Na+-Cl– and water transport; in contrast, patients with hypovolemic hyponatremia will often be hyperuricemic, due to a shared activation of proximal tubular Na+-Cl– and urate transport.
Common causes of SIAD include pulmonary disease (e.g., pneumonia, tuberculosis, pleural effusion) and central nervous system (CNS) diseases (e.g., tumor, subarachnoid hemorrhage, meningitis). SIAD also occurs with malignancies, most commonly with small-cell lung carcinoma (75% of malignancy-associated SIAD); ~10% of patients with this tumor will have a plasma Na+ concentration of <130 mM at presentation. SIAD is also a frequent complication of certain drugs, most commonly the selective serotonin reuptake inhibitors (SSRIs). Other drugs can potentiate the renal effect of AVP, without exerting direct effects on circulating AVP levels (Table 63-1).
Low Solute Intake and Hyponatremia Hyponatremia can occasionally occur in patients with a very low intake of dietary solutes. Classically, this occurs in alcoholics whose sole nutrient is beer, hence the diagnostic label of beer potomania; beer is very low in protein and salt content, containing only 1–2 mM of Na+. The syndrome has also been described in nonalcoholic patients with highly restricted solute intake due to nutrient-restricted diets, e.g., extreme vegetarian diets. Patients with hyponatremia due to low solute intake typically present with a very low urine osmolality (<100–200 mOsm/kg) with a urine Na+ concentration that is <10–20 mM. The fundamental abnormality is the inadequate dietary intake of solutes; the reduced urinary solute excretion limits water excretion such that hyponatremia ensues after relatively modest polydipsia. AVP levels have not been reported in patients with beer potomania but are expected to be suppressed or rapidly suppressible with saline hydration; this fits with the overly rapid correction in plasma Na+ concentration that can be seen with saline hydration. Resumption of a normal diet and/or saline hydration will also correct the causative deficit in urinary solute excretion, such that patients with beer potomania typically correct their plasma Na+ concentration promptly after admission to the hospital.
Clinical Features of Hyponatremia Hyponatremia induces generalized cellular swelling, a consequence of water movement down the osmotic gradient from the hypotonic ECF to the ICF. The symptoms of hyponatremia are primarily neurologic, reflecting the development of cerebral edema within a rigid skull. The initial CNS response to acute hyponatremia is an increase in interstitial pressure, leading to shunting of ECF and solutes from the interstitial space into the cerebrospinal fluid and then on into the systemic circulation. This is accompanied by an efflux of the major intracellular ions, Na+, K+, and Cl–, from brain cells. Acute hyponatremic encephalopathy ensues when these volume regulatory mechanisms are overwhelmed by a rapid decrease in tonicity, resulting in acute cerebral edema. Early symptoms can include nausea, headache, and vomiting. However, severe complications can rapidly evolve, including seizure activity, brainstem herniation, coma, and death. A key complication of acute hyponatremia is normocapneic or hypercapneic respiratory failure; the associated hypoxia may amplify the neurologic injury. Normocapneic respiratory failure in this setting is typically due to noncardiogenic, “neurogenic” pulmonary edema, with a normal pulmonary capillary wedge pressure.
Acute symptomatic hyponatremia is a medical emergency, occurring in a number of specific settings (Table 63-2). Women, particularly before menopause, are much more likely than men to develop encephalopathy and severe neurologic sequelae. Acute hyponatremia often has an iatrogenic component, e.g., when hypotonic intravenous fluids are given to postoperative patients with an increase in circulating AVP. Exercise-associated hyponatremia, an important clinical issue at marathons and other endurance events, has similarly been linked to both a “nonosmotic” increase in circulating AVP and excessive free water intake. The recreational drugs Molly and ecstasy, which share an active ingredient (MDMA, 3,4-methylenedioxymethamphetamine), cause a rapid and potent induction of both thirst and AVP, leading to severe acute hyponatremia.
CAUSES OF ACUTE HYPONATREMIA |
Abbreviations: MDMA, 3,4-methylenedioxymethamphetamine; TURP, transurethral resection of the prostate.
Persistent, chronic hyponatremia results in an efflux of organic osmolytes (creatine, betaine, glutamate, myoinositol, and taurine) from brain cells; this response reduces intracellular osmolality and the osmotic gradient favoring water entry. This reduction in intracellular osmolytes is largely complete within 48 h, the time period that clinically defines chronic hyponatremia; this temporal definition has considerable relevance for the treatment of hyponatremia (see below). The cellular response to chronic hyponatremia does not fully protect patients from symptoms, which can include vomiting, nausea, confusion, and seizures, usually at plasma Na+ concentration <125 mM. Even patients who are judged “asymptomatic” can manifest subtle gait and cognitive defects that reverse with correction of hyponatremia; notably, chronic “asymptomatic” hyponatremia increases the risk of falls. Chronic hyponatremia also increases the risk of bony fractures owing to the associated neurologic dysfunction and to a hyponatremia-associated reduction in bone density. Therefore, every attempt should be made to correct safely the plasma Na+ concentration in patients with chronic hyponatremia, even in the absence of overt symptoms (see the section on treatment of hyponatremia below).
The management of chronic hyponatremia is complicated significantly by the asymmetry of the cellular response to correction of plasma Na+ concentration. Specifically, the reaccumulation of organic osmolytes by brain cells is attenuated and delayed as osmolality increases after correction of hyponatremia, sometimes resulting in degenerative loss of oligodendrocytes and an osmotic demyelination syndrome (ODS). Overly rapid correction of hyponatremia (>8–10 mM in 24 h or 18 mM in 48 h) is also associated with a disruption in integrity of the blood-brain barrier, allowing the entry of immune mediators that may contribute to demyelination. The lesions of ODS classically affect the pons, a structure wherein the delay in the reaccumulation of osmotic osmolytes is particularly pronounced; clinically, patients with central pontine myelinolysis can present 1 or more days after overcorrection of hyponatremia with paraparesis or quadriparesis, dysphagia, dysarthria, diplopia, a “locked-in syndrome,” and/or loss of consciousness. Other regions of the brain can also be involved in ODS, most commonly in association with lesions of the pons but occasionally in isolation; in order of frequency, the lesions of extrapontine myelinolysis can occur in the cerebellum, lateral geniculate body, thalamus, putamen, and cerebral cortex or subcortex. Clinical presentation of ODS can, therefore, vary as a function of the extent and localization of extrapontine myelinolysis, with the reported development of ataxia, mutism, parkinsonism, dystonia, and catatonia. Relowering of plasma Na+ concentration after overly rapid correction can prevent or attenuate ODS (see the section on treatment of hyponatremia below). However, even appropriately slow correction can be associated with ODS, particularly in patients with additional risk factors; these include alcoholism, malnutrition, hypokalemia, and liver transplantation.
Diagnostic Evaluation of Hyponatremia Clinical assessment of hyponatremic patients should focus on the underlying cause; a detailed drug history is particularly crucial (Table 63-1). A careful clinical assessment of volume status is obligatory for the classical diagnostic approach to hyponatremia (Fig. 63-5). Hyponatremia is frequently multifactorial, particularly when severe; clinical evaluation should consider all the possible causes for excessive circulating AVP, including volume status, drugs, and the presence of nausea and/or pain. Radiologic imaging may also be appropriate to assess whether patients have a pulmonary or CNS cause for hyponatremia. A screening chest x-ray may fail to detect a small-cell carcinoma of the lung; computed tomography (CT) scanning of the thorax should be considered in patients at high risk for this tumor (e.g., patients with a smoking history).
Laboratory investigation should include a measurement of serum osmolality to exclude pseudohyponatremia, which is defined as the coexistence of hyponatremia with a normal or increased plasma tonicity. Most clinical laboratories measure plasma Na+ concentration by testing diluted samples with automated ion-sensitive electrodes, correcting for this dilution by assuming that plasma is 93% water. This correction factor can be inaccurate in patients with pseudohyponatremia due to extreme hyperlipidemia and/or hyperproteinemia, in whom serum lipid or protein makes up a greater percentage of plasma volume. The measured osmolality should also be converted to the effective osmolality (tonicity) by subtracting the measured concentration of urea (divided by 2.8, if in mg/dL); patients with hyponatremia have an effective osmolality of <275 mOsm/kg.
Elevated BUN and creatinine in routine chemistries can also indicate renal dysfunction as a potential cause of hyponatremia, whereas hyperkalemia may suggest adrenal insufficiency or hypoaldosteronism. Serum glucose should also be measured; plasma Na+ concentration falls by ~1.6–2.4 mM for every 100-mg/dL increase in glucose, due to glucose-induced water efflux from cells; this “true” hyponatremia resolves after correction of hyperglycemia. Measurement of serum uric acid should also be performed; whereas patients with SIAD-type physiology will typically be hypouricemic (serum uric acid <4 mg/dL), volume-depleted patients will often be hyperuricemic. In the appropriate clinical setting, thyroid, adrenal, and pituitary function should also be tested; hypothyroidism and secondary adrenal failure due to pituitary insufficiency are important causes of euvolemic hyponatremia, whereas primary adrenal failure causes hypovolemic hyponatremia. A cosyntropin stimulation test is necessary to assess for primary adrenal insufficiency.
Urine electrolytes and osmolality are crucial tests in the initial evaluation of hyponatremia. A urine Na+ concentration <20–30 mM is consistent with hypovolemic hyponatremia, in the clinical absence of a hypervolemic, Na+-avid syndrome such as CHF (Fig. 63-5). In contrast, patients with SIAD will typically excrete urine with an Na+ concentration that is >30 mM. However, there can be substantial overlap in urine Na+ concentration values in patients with SIAD and hypovolemic hyponatremia, particularly in the elderly; the ultimate “gold standard” for the diagnosis of hypovolemic hyponatremia is the demonstration that plasma Na+ concentration corrects after hydration with normal saline. Patients with thiazide-associated hyponatremia may also present with higher than expected urine Na+ concentration and other findings suggestive of SIAD; one should defer making a diagnosis of SIAD in these patients until 1–2 weeks after discontinuing the thiazide. A urine osmolality <100 mOsm/kg is suggestive of polydipsia; urine osmolality >400 mOsm/kg indicates that AVP excess is playing a more dominant role, whereas intermediate values are more consistent with multifactorial pathophysiology (e.g., AVP excess with a significant component of polydipsia). Patients with hyponatremia due to decreased solute intake (beer potomania) typically have urine Na+ concentration <20 mM and urine osmolality in the range of <100 to the low 200s. Finally, the measurement of urine K+ concentration is required to calculate the urine-to-plasma electrolyte ratio, which is useful to predict the response to fluid restriction (see the section on treatment of hyponatremia below).
HYPONATREMIA |
Three major considerations guide the therapy of hyponatremia. First, the presence and/or severity of symptoms determine the urgency and goals of therapy. Patients with acute hyponatremia (Table 63-2) present with symptoms that can range from headache, nausea, and/or vomiting, to seizures, obtundation, and central herniation; patients with chronic hyponatremia, present for >48 h, are less likely to have severe symptoms. Second, patients with chronic hyponatremia are at risk for ODS if plasma Na+ concentration is corrected by >8–10 mM within the first 24 h and/or by >18 mM within the first 48 h. Third, the response to interventions such as hypertonic saline, isotonic saline, or AVP antagonists can be highly unpredictable, such that frequent monitoring of plasma Na+ concentration during corrective therapy is imperative.
Once the urgency in correcting the plasma Na+ concentration has been established and appropriate therapy instituted, the focus should be on treatment or withdrawal of the underlying cause. Patients with euvolemic hyponatremia due to SIAD, hypothyroidism, or secondary adrenal failure will respond to successful treatment of the underlying cause, with an increase in plasma Na+ concentration. However, not all causes of SIAD are immediately reversible, necessitating pharmacologic therapy to increase the plasma Na+ concentration (see below). Hypovolemic hyponatremia will respond to intravenous hydration with isotonic normal saline, with a rapid reduction in circulating AVP and a brisk water diuresis; it may be necessary to reduce the rate of correction if the history suggests that hyponatremia has been chronic, i.e., present for more than 48 h (see below). Hypervolemic hyponatremia due to CHF will often respond to improved therapy of the underlying cardiomyopathy, e.g., following the institution or intensification of angiotensin-converting enzyme (ACE) inhibition. Finally, patients with hyponatremia due to beer potomania and low solute intake will respond very rapidly to intravenous saline and the resumption of a normal diet. Notably, patients with beer potomania have a very high risk of developing ODS, due to the associated hypokalemia, alcoholism, malnutrition, and high risk of overcorrecting the plasma Na+ concentration.
Water deprivation has long been a cornerstone of the therapy of chronic hyponatremia. However, patients who are excreting minimal electrolyte-free water will require aggressive fluid restriction; this can be very difficult for patients with SIAD to tolerate, given that their thirst is also inappropriately stimulated. The urine-to-plasma electrolyte ratio (urinary [Na+] + [K+]/plasma [Na+]) can be exploited as a quick indicator of electrolyte-free water excretion (Table 63-3); patients with a ratio of >1 should be more aggressively restricted (<500 mL/d), those with a ratio of ~1 should be restricted to 500–700 mL/d, and those with a ratio <1 should be restricted to <1 L/d. In hypokalemic patients, potassium replacement will serve to increase plasma Na+ concentration, given that the plasma Na+ concentration is a functional of both exchangeable Na+ and exchangeable K+ divided by total-body water; a corollary is that aggressive repletion of K+ has the potential to overcorrect the plasma Na+ concentration even in the absence of hypertonic saline. Plasma Na+ concentration will also tend to respond to an increase in dietary solute intake, which increases the ability to excrete free water; however, the use of oral urea and/or salt tablets for this purpose is generally not practical or well tolerated.
MANAGEMENT OF HYPERNATREMIA |
Ongoing Water Losses
4. Calculate free-water clearance, CeH2O:
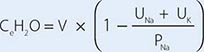
where V is urinary volume, UNa is urinary [Na+], UK is urinary [K+], and PNa is plasma [Na+]
Patients in whom therapy with fluid restriction, potassium replacement, and/or increased solute intake fails may merit pharmacologic therapy to increase their plasma Na+ concentration. Many patients with SIAD respond to combined therapy with oral furosemide, 20 mg twice a day (higher doses may be necessary in renal insufficiency), and oral salt tablets; furosemide serves to inhibit the renal countercurrent mechanism and blunt urinary concentrating ability, whereas the salt tablets counteract diuretic-associated natriuresis. Demeclocycline is a potent inhibitor of principal cells and can be used in patients whose Na levels do not increase in response to furosemide and salt tablets. However, this agent can be associated with a reduction in GFR, due to excessive natriuresis and/or direct renal toxicity; it should be avoided in cirrhotic patients in particular, who are at higher risk of nephrotoxicity due to drug accumulation.
AVP antagonists (vaptans) are highly effective in SIAD and in hypervolemic hyponatremia due to heart failure or cirrhosis, reliably increasing plasma Na+ concentration due to their “aquaretic” effects (augmentation of free water clearance). Most of these agents specifically antagonize the V2 AVP receptor; tolvaptan is currently the only oral V2 antagonist to be approved by the U.S. Food and Drug Administration. Conivaptan, the only available intravenous vaptan, is a mixed V1A/V2 antagonist, with a modest risk of hypotension due to V1A receptor inhibition. Therapy with vaptans must be initiated in a hospital setting, with a liberalization of fluid restriction (>2 L/d) and close monitoring of plasma Na+ concentration. Although approved for the management of all but hypovolemic hyponatremia and acute hyponatremia, the clinical indications for these agents are not completely clear. Oral tolvaptan is perhaps most appropriate for the management of significant and persistent SIAD (e.g., in small-cell lung carcinoma) that has not responded to water restriction and/or oral furosemide and salt tablets. Abnormalities in liver function tests have been reported with chronic tolvaptan therapy; hence, the use of this agent should be restricted to <1–2 months.
Treatment of acute symptomatic hyponatremia should include hypertonic 3% saline (513 mM) to acutely increase plasma Na+ concentration by 1–2 mM/h to a total of 4–6 mM; this modest increase is typically sufficient to alleviate severe acute symptoms, after which corrective guidelines for chronic hyponatremia are appropriate (see below). A number of equations have been developed to estimate the required rate of hypertonic saline, which has an Na+-Cl– concentration of 513 mM. The traditional approach is to calculate an Na+ deficit, where the Na+ deficit = 0.6 × body weight × (target plasma Na+ concentration – starting plasma Na+ concentration), followed by a calculation of the required rate. Regardless of the method used to determine the rate of administration, the increase in plasma Na+ concentration can be highly unpredictable during treatment with hypertonic saline, due to rapid changes in the underlying physiology; plasma Na+ concentration should be monitored every 2–4 h during treatment, with appropriate changes in therapy based on the observed rate of change. The administration of supplemental oxygen and ventilatory support is also critical in acute hyponatremia, in the event that patients develop acute pulmonary edema or hypercapneic respiratory failure. Intravenous loop diuretics will help treat acute pulmonary edema and will also increase free water excretion, by interfering with the renal countercurrent multiplication system. AVP antagonists do not have an approved role in the management of acute hyponatremia.
The rate of correction should be comparatively slow in chronic hyponatremia (<8–10 mM in the first 24 h and <18 mM in the first 48 h), so as to avoid ODS; lower target rates are appropriate in patients at particular risk for ODS, such as alcoholics or hypokalemic patients. Overcorrection of the plasma Na+ concentration can occur when AVP levels rapidly normalize, for example following the treatment of patients with chronic hypovolemic hyponatremia with intravenous saline or following glucocorticoid replacement of patients with hypopituitarism and secondary adrenal failure. Approximately 10% of patients treated with vaptans will overcorrect; the risk is increased if water intake is not liberalized. In the event that the plasma Na+ concentration overcorrects following therapy, be it with hypertonic saline, isotonic saline, or a vaptan, hyponatremia can be safely reinduced or stabilized by the administration of the AVP agonist desmopressin acetate (DDAVP) and/or the administration of free water, typically intravenous D5W; the goal is to prevent or reverse the development of ODS. Alternatively, the treatment of patients with marked hyponatremia can be initiated with the twice-daily administration of DDAVP to maintain constant AVP bioactivity, combined with the administration of hypertonic saline to slowly correct the serum sodium in a more controlled fashion, thus reducing upfront the risk of overcorrection.
HYPERNATREMIA
Etiology Hypernatremia is defined as an increase in the plasma Na+ concentration to >145 mM. Considerably less common than hyponatremia, hypernatremia is nonetheless associated with mortality rates of as high as 40–60%, mostly due to the severity of the associated underlying disease processes. Hypernatremia is usually the result of a combined water and electrolyte deficit, with losses of H2O in excess of Na+. Less frequently, the ingestion or iatrogenic administration of excess Na+ can be causative, for example after IV administration of excessive hypertonic Na+-Cl– or Na+-HCO3– (Fig. 63-6).
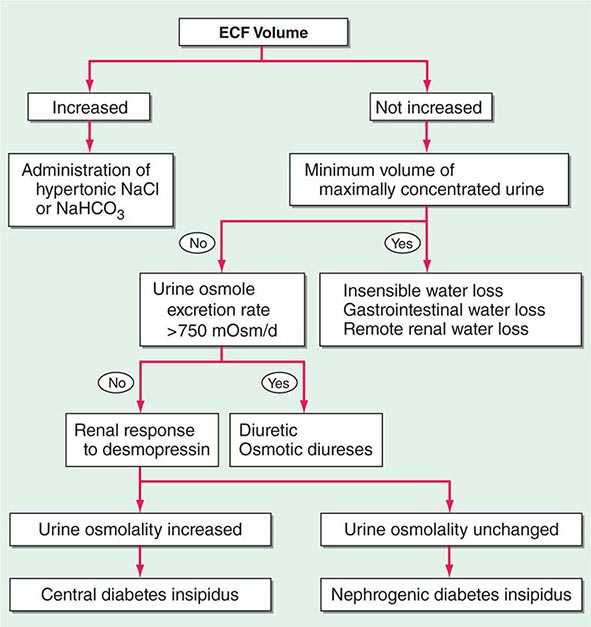
FIGURE 63-6 The diagnostic approach to hypernatremia. ECF, extracellular fluid.
Elderly individuals with reduced thirst and/or diminished access to fluids are at the highest risk of developing hypernatremia. Patients with hypernatremia may rarely have a central defect in hypothalamic osmoreceptor function, with a mixture of both decreased thirst and reduced AVP secretion. Causes of this adipsic diabetes insipidus include primary or metastatic tumor, occlusion or ligation of the anterior communicating artery, trauma, hydrocephalus, and inflammation.
Hypernatremia can develop following the loss of water via both renal and nonrenal routes. Insensible losses of water may increase in the setting of fever, exercise, heat exposure, severe burns, or mechanical ventilation. Diarrhea is, in turn, the most common gastrointestinal cause of hypernatremia. Notably, osmotic diarrhea and viral gastroenteritides typically generate stools with Na+ and K+ <100 mM, thus leading to water loss and hypernatremia; in contrast, secretory diarrhea typically results in isotonic stool and thus hypovolemia with or without hypovolemic hyponatremia.
Common causes of renal water loss include osmotic diuresis secondary to hyperglycemia, excess urea, postobstructive diuresis, or mannitol; these disorders share an increase in urinary solute excretion and urinary osmolality (see “Diagnostic Approach,” below). Hypernatremia due to a water diuresis occurs in central or nephrogenic diabetes insipidus (DI).
Nephrogenic DI (NDI) is characterized by renal resistance to AVP, which can be partial or complete (see “Diagnostic Approach,” below). Genetic causes include loss-of-function mutations in the X-linked V2 receptor; mutations in the AVP-responsive aquaporin-2 water channel can cause autosomal recessive and autosomal dominant NDI, whereas recessive deficiency of the aquaporin-1 water channel causes a more modest concentrating defect (Fig. 63-2). Hypercalcemia can also cause polyuria and NDI; calcium signals directly through the calcium-sensing receptor to downregulate Na+, K+, and Cl– transport by the TALH and water transport in principal cells, thus reducing renal concentrating ability in hypercalcemia. Another common acquired cause of NDI is hypokalemia, which inhibits the renal response to AVP and downregulates aquaporin-2 expression. Several drugs can cause acquired NDI, in particular lithium, ifosfamide, and several antiviral agents. Lithium causes NDI by multiple mechanisms, including direct inhibition of renal glycogen synthase kinase-3 (GSK3), a kinase thought to be the pharmacologic target of lithium in bipolar disease; GSK3 is required for the response of principal cells to AVP. The entry of lithium through the amiloride-sensitive Na+ channel ENaC (Fig. 63-4) is required for the effect of the drug on principal cells, such that combined therapy within lithium and amiloride can mitigate lithium-associated NDI. However, lithium causes chronic tubulointerstitial scarring and chronic kidney disease after prolonged therapy, such that patients may have a persistent NDI long after stopping the drug, with a reduced therapeutic benefit from amiloride.
Finally, gestational DI is a rare complication of late-term pregnancy wherein increased activity of a circulating placental protease with “vasopressinase” activity leads to reduced circulating AVP and polyuria, often accompanied by hypernatremia. DDAVP is an effective therapy for this syndrome, given its resistance to the vasopressinase enzyme.
Clinical Features Hypernatremia increases osmolality of the ECF, generating an osmotic gradient between the ECF and ICF, an efflux of intracellular water, and cellular shrinkage. As in hyponatremia, the symptoms of hypernatremia are predominantly neurologic. Altered mental status is the most frequent manifestation, ranging from mild confusion and lethargy to deep coma. The sudden shrinkage of brain cells in acute hypernatremia may lead to parenchymal or subarachnoid hemorrhages and/or subdural hematomas; however, these vascular complications are primarily encountered in pediatric and neonatal patients. Osmotic damage to muscle membranes can also lead to hypernatremic rhabdomyolysis. Brain cells accommodate to a chronic increase in ECF osmolality (>48 h) by activating membrane transporters that mediate influx and intracellular accumulation of organic osmolytes (creatine, betaine, glutamate, myoinositol, and taurine); this results in an increase in ICF water and normalization of brain parenchymal volume. In consequence, patients with chronic hypernatremia are less likely to develop severe neurologic compromise. However, the cellular response to chronic hypernatremia predisposes these patients to the development of cerebral edema and seizures during overly rapid hydration (overcorrection of plasma Na+ concentration by >10 mM/d).
Diagnostic Approach The history should focus on the presence or absence of thirst, polyuria, and/or an extrarenal source for water loss, such as diarrhea. The physical examination should include a detailed neurologic exam and an assessment of the ECFV; patients with a particularly large water deficit and/or a combined deficit in electrolytes and water may be hypovolemic, with reduced JVP and orthostasis. Accurate documentation of daily fluid intake and daily urine output is also critical for the diagnosis and management of hypernatremia.
Laboratory investigation should include a measurement of serum and urine osmolality, in addition to urine electrolytes. The appropriate response to hypernatremia and a serum osmolality >295 mOsm/kg is an increase in circulating AVP and the excretion of low volumes (<500 mL/d) of maximally concentrated urine, i.e., urine with osmolality >800 mOsm/kg; should this be the case, then an extrarenal source of water loss is primarily responsible for the generation of hypernatremia. Many patients with hypernatremia are polyuric; should an osmotic diuresis be responsible, with excessive excretion of Na+-Cl–, glucose, and/or urea, then daily solute excretion will be >750–1000 mOsm/d (>15 mOsm/kg body water per day) (Fig. 63-6). More commonly, patients with hypernatremia and polyuria will have a predominant water diuresis, with excessive excretion of hypotonic, dilute urine.
Adequate differentiation between nephrogenic and central causes of DI requires the measurement of the response in urinary osmolality to DDAVP, combined with measurement of circulating AVP in the setting of hypertonicity. By definition, patients with baseline hypernatremia are hypertonic, with an adequate stimulus for AVP by the posterior pituitary. Therefore, in contrast to polyuric patients with a normal or reduced baseline plasma Na+ concentration and osmolality, a water deprivation test (Chap. 61) is unnecessary in hypernatremia; indeed, water deprivation is absolutely contraindicated in this setting, given the risk for worsening the hypernatremia. Patients with NDI will fail to respond to DDAVP, with a urine osmolality that increases by <50% or <150 mOsm/kg from baseline, in combination with a normal or high circulating AVP level; patients with central DI will respond to DDAVP, with a reduced circulating AVP. Patients may exhibit a partial response to DDAVP, with a >50% rise in urine osmolality that nonetheless fails to reach 800 mOsm/kg; the level of circulating AVP will help differentiate the underlying cause, i.e., NDI versus central DI. In pregnant patients, AVP assays should be drawn in tubes containing the protease inhibitor 1,10-phenanthroline, to prevent in vitro degradation of AVP by placental vasopressinase.
For patients with hypernatremia due to renal loss of water, it is critical to quantify ongoing daily losses using the calculated electrolyte-free water clearance, in addition to calculation of the baseline water deficit (the relevant formulas are discussed in Table 63-3). This requires daily measurement of urine electrolytes, combined with accurate measurement of daily urine volume.
TREATMENT | HYPERNATREMIA |
The underlying cause of hypernatremia should be withdrawn or corrected, be it drugs, hyperglycemia, hypercalcemia, hypokalemia, or diarrhea. The approach to the correction of hypernatremia is outlined in Table 63-3. It is imperative to correct hypernatremia slowly to avoid cerebral edema, typically replacing the calculated free water deficit over 48 h. Notably, the plasma Na+ concentration should be corrected by no more than 10 mM/d, which may take longer than 48 h in patients with severe hypernatremia (>160 mM). A rare exception is patients with acute hypernatremia (<48 h) due to sodium loading, who can safely be corrected rapidly at a rate of 1 mM/h.
Water should ideally be administered by mouth or by nasogastric tube, as the most direct way to provide free water, i.e., water without electrolytes. Alternatively, patients can receive free water in dextrose-containing IV solutions, such as 5% dextrose (D5W); blood glucose should be monitored in case hyperglycemia occurs. Depending on the history, blood pressure, or clinical volume status, it may be appropriate to initially treat with hypotonic saline solutions (1/4 or 1/2 normal saline); normal saline is usually inappropriate in the absence of very severe hypernatremia, where normal saline is proportionally more hypotonic relative to plasma, or frank hypotension. Calculation of urinary electrolyte-free water clearance (Table 63-3) is required to estimate daily, ongoing loss of free water in patients with NDI or central DI, which should be replenished daily.
Additional therapy may be feasible in specific cases. Patients with central DI should respond to the administration of intravenous, intranasal, or oral DDAVP. Patients with NDI due to lithium may reduce their polyuria with amiloride (2.5–10 mg/d), which decreases entry of lithium into principal cells by inhibiting ENaC (see above); in practice, however, most patients with lithium-associated DI are able to compensate for their polyuria by simply increasing their daily water intake. Thiazides may reduce polyuria due to NDI, ostensibly by inducing hypovolemia and increasing proximal tubular water reabsorption. Occasionally, nonsteroidal anti-inflammatory drugs (NSAIDs) have been used to treat polyuria associated with NDI, reducing the negative effect of intrarenal prostaglandins on urinary concentrating mechanisms; however, this assumes the risks of NSAID-associated gastric and/or renal toxicity. Furthermore, it must be emphasized that thiazides, amiloride, and NSAIDs are only appropriate for chronic management of polyuria from NDI and have no role in the acute management of associated hypernatremia, where the focus is on replacing free water deficits and ongoing free water loss.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


