significant tissue response. Intraocular foreign bodies containing iron are particularly toxic to the retina, and they may cause a diffuse deposition of iron throughout the eye (13). Copper-rich foreign bodies incite a significant intraocular acute inflammatory reaction (14). Other metals such as lead, zinc, nickel, aluminum, and mercury may also evoke intraocular inflammation.
TABLE 24.1 Benign Intraocular Tumors | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||
TABLE 24.2 Causes of Intraocular Granulomatous Inflammation | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||
optic nerve is characterized by an accumulation of hyaluronic acid that stains positively with the Hale colloidal iron technique or the Alcian blue stain (Schnabel cavernous atrophy). An examination of a glaucomatous eye may also disclose the cause of congenital or secondary glaucoma. For example, fibrocollagenous adhesions may be present between the posterior surface of the peripheral cornea and the iris (peripheral anterior synechiae), and this may have obstructed the aqueous humor outflow.
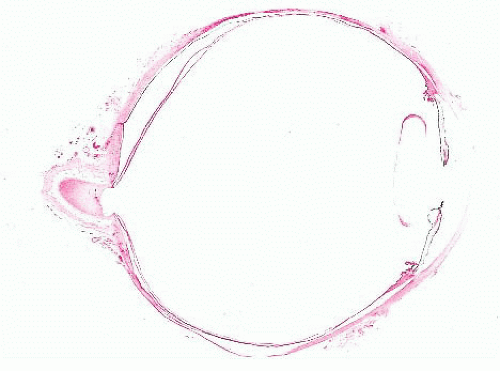 FIGURE 24.1 Marked cupping of the optic disc. This eye was surgically excised (enucleated) because of glaucoma. |
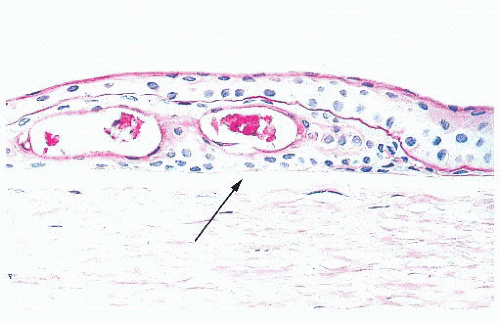 FIGURE 24.2 Basement membrane material within the epithelial layer of the cornea. Also present are two intraepithelial cysts. Note that Bowman layer (arrow) does not react positively with the periodic acid-Schiff stain. |
associated with inflammation. With aging, corneal endothelial cells diminish in number, and Descemet membrane thickens. A diffuse, irregular thickening of Descemet membrane accompanies some long-standing degenerative changes of the endothelial layer. Fibrous retrocorneal membranes between the Descemet membrane and the corneal endothelium may follow grafts or various inflammatory processes of the cornea (46).
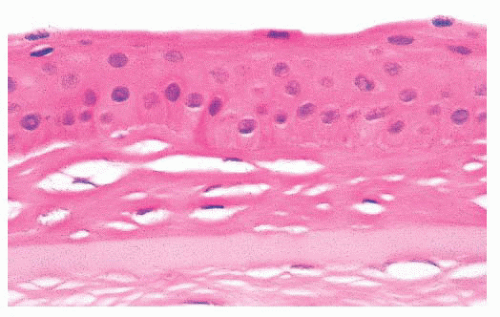 FIGURE 24.3 Collagenous tissue between the corneal epithelium and Bowman layer (pannus). |
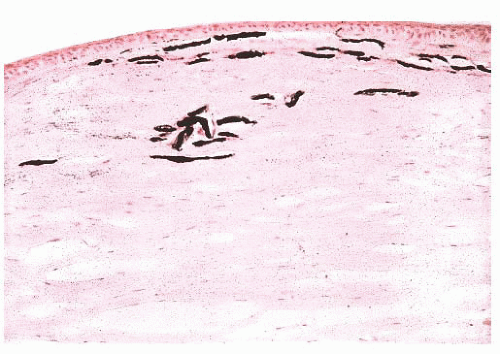 FIGURE 24.4 Calcific band keratopathy. Linear deposits of calcium phosphate are present within the superficial corneal stroma (von Kossa stain). |
to specific chromosomes, and the genes have been identified in many of them. Different genetic mutations have been related with CDs development. Most frequently involved genes are TGFBI, CHST6, KRT3, KRT12, PIP 5k3, SLC 4AIII, TATICSt2, and UBIADI.
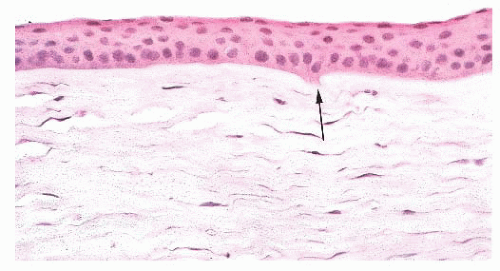 FIGURE 24.5 Keratoconus. Focal disruptions of Bowman layer (arrow) are common. |
TABLE 24.3 Significant Features and Predominant Corneal Layer Affected in Some Corneal Dystrophies | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
TABLE 24.4 Stains Used and Patterns for Histopathologic Diagnosis of the Stromal Corneal Dystrophies | |||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||
 FIGURE 24.6 Fuchs corneal dystrophy. Excrescences form over the peripheral and central part of Descemet membrane. The corneal endothelial cells are diminished in number, and Descemet membrane is also often abnormally thickened. |
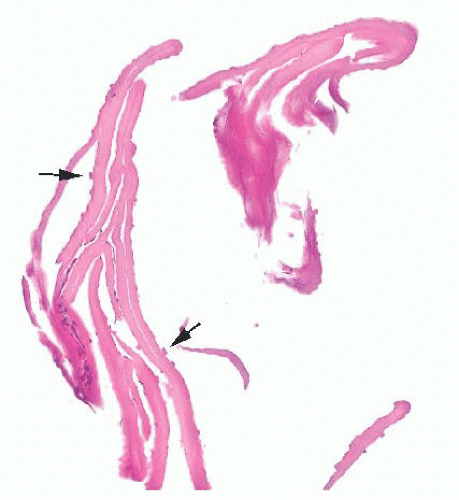 FIGURE 24.7 Fuchs corneal dystrophy. Descemet membrane and endothelium showing guttae (arrows). |
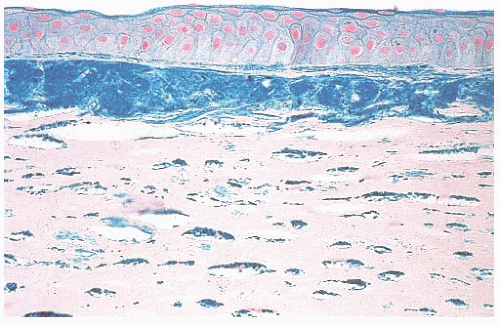 FIGURE 24.8 Macular corneal dystrophy. Extracellular stromal deposits of glycosaminoglycans are found. Similar material is also present within the corneal fibroblasts (keratocytes) (Hale colloidal iron). |
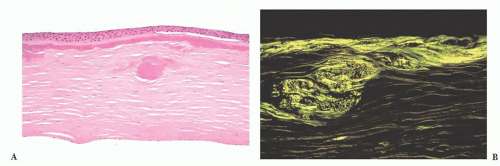 FIGURE 24.9 Lattice corneal dystrophy. (A) Amyloid accumulation is shown within the corneal stroma in a variant of lattice corneal dystrophy type 1. In this photomicrograph, amyloid is evident immediately beneath the Bowman layer and within the anterior corneal stroma. (B) The amyloid is birefringent and exhibits apple green dichroism after being stained with Congo red. |
causative microorganism is often difficult to detect in tissue sections without the aid of special stains. Colonies of some bacteria, such as Streptococcus viridans, may produce crystalline-like stromal opacities in the absence of an inflammatory cell infiltrate (“infectious pseudocrystalline keratopathy”) (81).
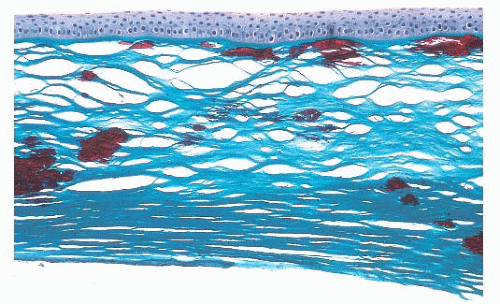 FIGURE 24.10 Granular corneal dystrophy. Abnormal collections of mutated transforming growth factor-β-induced protein accumulate within the corneal stroma. This material appears bright red with the Masson trichrome stain. |
TABLE 24.5 Some Infectious Organisms in Keratitis | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||
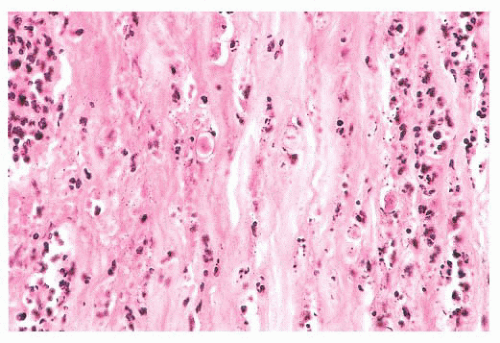 FIGURE 24.11 Acanthamoebic keratitis. Amebic keratitis is characterized by numerous stromal polymorphonuclear leukocytes and necrotic tissue. The amebae are evident within the affected tissue. |
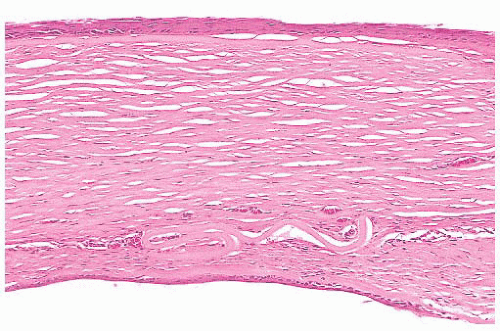 FIGURE 24.12 Transformation of corneal endothelium to stratified squamous epithelium. Epithelium derived from the conjunctiva has entered the eye through a traumatic wound. This ingrowth of aberrant squamous epithelium may extend along the posterior surface of the cornea and into the anterior chamber angle. Collagenous tissue is often present between remnants of Descemet membrane and the ectopic squamous epithelium. Neovascularization of the posterior corneal stroma is also evident in this photomicrograph. Histochemical markers for cytokeratins are useful in selected cases of epithelial ingrowth. |
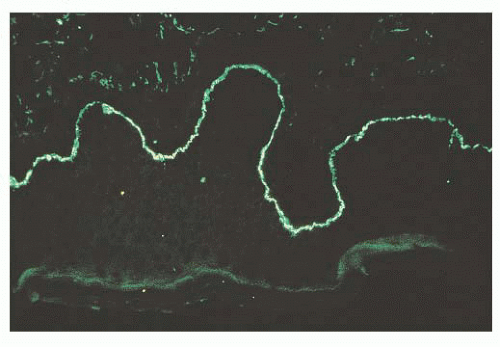 FIGURE 24.13 Ocular cicatricial pemphigoid. Linear deposits of immunoglobulin G (IgG) (pictured), as well as immunoglobulin A (IgA), accumulate along the basement membrane of the conjunctival epithelium. |
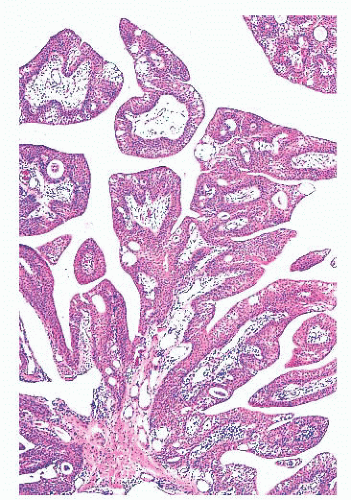 FIGURE 24.14 Conjunctival papillomas. Lobules of squamous epithelium surrounding a fibrovascular core characterize these papillomas histologically. |
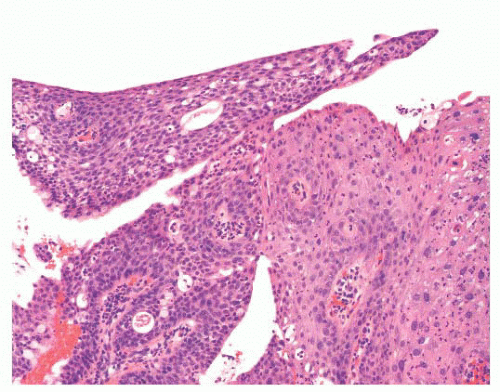 FIGURE 24.15 Conjunctival papilloma with dysplasia. In rare instances, a conjunctival papilloma can exhibit epithelial dysplasia (right side of the picture). In this particular case, the papilloma is a low-grade intraepithelial dysplasia. |
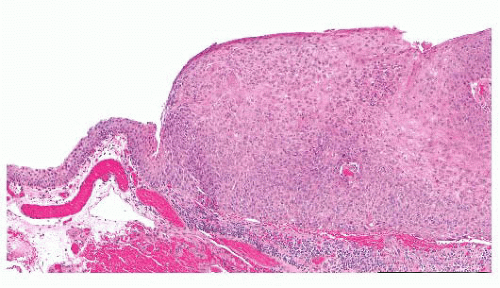 FIGURE 24.16 Conjunctival intraepithelial neoplasia. The atypical epithelial cells are present in the entire thickness of the epithelium, characterizing carcinoma in situ. |
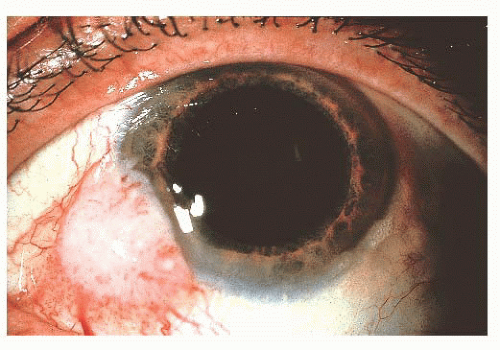 FIGURE 24.17 Squamous cell carcinoma of the conjunctiva. These most often arise at the corneoscleral limbus. |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


