Neoplasm or not neoplasm: This hypercellular lesion is composed of pleomorphic cells with atypical nuclei, which
indicate neoplasm. If the lesion were not neoplastic, other etiologic categories, including infectious, inflammatory, toxic-metabolic, traumatic, vascular, developmental, and degenerative, would be considered.
TABLE 10.1 Indications for Intraoperative Consultation at Biopsy
Lesion requires proper surgical sampling of tissue for diagnosis and grading
Lesion may require special tissue processing
Culture for microorganisms
Fixation for electron microscopy
Fixation for immunohistochemistry
Touch preparation
Other special processing
Diagnosis affects immediate surgical procedure
TABLE 10.2 Surgery Directed Toward a Neurologic Symptom or Specific Disease
CONFIRMATORY FEATURES OF SUSPECTED DISEASE
Symptom/Suspected Disease
Structures
Reactant
Locationsa
Herpes simplex encephalitis
Encephalitis (Table 10.3), Cowdry A amphophilic nuclear inclusions of 90- to 100-nm “target” capsids
HSV antigen
Temporal or basilar frontal lobe, CNS; frequently bilateral
Toxoplasmosis
Necrosis containing 3- to 5-nm tachyzoites; (cysts); (inflammation)b
Toxoplasma antigen
CNS, frequent multiple lesions
Progressive multifocal leukoencephalopathy
Demyelination, bizarre, glial, amphophilic nuclear inclusions of 15- to 25-nm or 30- to 40-nm diameter
JC/SV40 antigen, myelin, neurofilament
Cerebral white matter, CNS
Dementia/Creutzfeldt-Jakob disease
Cytoplasmic vacuoles indenting nuclei, gliosis (Table 10.3)
GFAP, prion (DANGER)
Bilateral cerebral cortex, gray matter
Small vessel disease
Vasculitis or arterial sclerosis or congophilic angiopathy
Amyloid, iron
Cerebrum, CNS; frequent multiple lesions
Dementia/Alzheimer disease
Argyrophilic plaques; neurofibrillary tangles of bihelical filaments
Neurofilament, tau
Bilateral cerebral cortex
Demyelination
Loss of myelin, gitter cells, with or without axonal preservation
Myelin, neurofilament
Cerebral white matter, CNS
Epilepsy
Low-grade glioma or ganglioglioma (Table 10.10), or gliosis (Tables 10.3 and 10.18), or vascular malformation
GFAP, neurofilament, synaptophysin, iron
Temporal lobe, cerebral cortex
a Most common or most specific location is listed first.
b Parentheses around a differential feature indicate an uncommon feature very useful in differential diagnosis when found.
CNS, central nervous system; GFAP, glial fibrillary acidic protein; HSV, herpes simplex virus; JC, JC virus; SV, simian virus.
TABLE 10.3 Differential Features of Cells Infiltrating Central Nervous Parenchyma
DIFFERENTIAL FEATURES
Diagnosis
Structures
Reactant
Locationsa
Gliosisb
Cells are fibrillar, uncrowded; round or oval nuclei
GFAP in glial filament
CNS
Macrophages
Cells and nuclei are round to elongated; cell content reflects injury
CD68; α-ACT
CNS, meninges
Encephalitis and/or cerebritis
Perivascular mixture of inflammatory cells
CD3, CD20, LCA, κ and λ Ig, α-ACT; CD68, microorganism
CNS gray matter, CNS
Hemorrhage
Red blood cells or macrophages with hemosiderin
Fibrin, iron
Deep cerebrum, cerebellum, CNS
Margin of gliomasc
Cells are fibrillar; angular nuclei indent each other; (mitoses)c,d
GFAP
CNS
Lymphoma
Perivascular, noncohesive small, round cells
CD3, CD20, LCA, κ and λ Ig
Deep cerebrum, CNS; meninges
a Most common or most specific location is listed first.
b Nonspecific reaction to injury.
c Suspicion of margin of glioma on frozen section should be followed by a request for another more central biopsy. Mitoses suggest margin of a high-grade glioma.
d Parentheses around a differential feature indicate an uncommon feature that is very useful in differential diagnosis when it is found.
α-ACT, α-antichymotrypsin macrophage marker; CNS, central nervous system; GFAP, glial fibrillary acidic protein; Ig, immunoglobulin; LCA, leukocyte common antigen (CD45/CD45RB).
TABLE 10.4 Viral Inclusions in the Central Nervous System
Nucleus
Cytoplasm
Neurons
Herpes simplex and zoster
+
−
Rabies
−
+
SSPE (measles)
+
−
Oligodendrocytes
PML (JC virus)
+
−
SSPE (measles)
+
−
Various cells (endothelial, ependymal, glial)
Cytomegalovirus
+
+
PML, progressive multifocal leukoencephalopathy; SSPE, subacute sclerosing panencephalitis.
TABLE 10.5 Causes to Consider in Intracranial Hematoma
Traumatic
Cerebral contusion
Epidural or subdural hematoma
Infectious and inflammatory
Vasculitis
Fungus (mycotic aneurysm)
Developmental vascular
Vascular malformation (arteriovenous malformation, cavernous angioma)
Aneurysm
Hematologic
Thrombocytopenia
Sickle cell anemia
Neoplastic
Primary (high-grade glioma)
Metastatic (e.g., melanoma, renal cell carcinoma)
Leukemia (chloroma in acute myelogenous leukemia)
Vascular
Amyloid angiopathy
Hypertension
Conversion of ischemic infarct
Toxic-metabolic: anticoagulation related
TABLE 10.6 Vascular Malformations
Malformationa
Vessels
Neuropil
Gliosis and Hemosiderin-Laden Macrophages
Arteriovenous malformation
Arteries, veins, arterialized veins
Absent, except around feeding vessels
Presentb
Cavernous angioma
Compact cluster thin-walled to thickwalled vessels, mineralization
Absent usually
Presentb
Capillary telangiectasia
Capillaries, thin-walled
Admixed
Absent
Venous malformation
Veins, often dilated
Admixed
Absent
Varix
Vein, dilated (vein of Galen)
Absent
Absent unless ruptured
a Location: Each can be found anywhere in the central nervous system; capillary telangiectasias often in brainstem and venous malformations in the spinal leptomeninges.
b Gliosis and hemosiderin-laden macrophages are consistent with leakage; they serve as a seizure focus, and they signal a tendency for spontaneous hemorrhage.
TABLE 10.7 Relative Frequency of Most Common Pediatric and Adult Brain Neoplasms
CHILDREN
ADULTS
Location
Diagnosis and Frequency
Location
Diagnosis and Frequency
Anterior fossa
Miscellaneous
33%
Anterior fossa
Gliomas (cerebrum)a
33%
Meningiomas (dura)
13%
Metastases (cerebrum)
12%
Pituitary adenomas (sella)
5%
Miscellaneous
4%
Total
67%
Posterior fossa
Astrocytomas (cerebellum)a
26%
Medulloblastomas (cerebellum)
24%
Ependymomas (fourth ventricle)
14%
Posterior fossa
Schwannomas (eighth cranial nerve)
8%
Miscellaneous
3%
Miscellaneous
25%
Total
67%
Total
33%
a Most common site in parentheses. See entry in text for other locations.
TABLE 10.8 World Health Organization Criteria for Grading of Astrocytomas
Grade
Nomenclature
Histologic Features
1
Pilocytic
Circumscribed; biphasic: bipolar piloid cells and multipolar cells; microcysts, Rosenthal fibers, and granular bodies; may or may not have rare mitotic figures, vascular proliferation, or focal necrosis
2
Diffuse
Moderate hypercellularity of monotonous cells; mild nuclear atypia; no or minimal mitotic activity
3
Anaplastic
Increased cellularity and diffuse infiltration; increased nuclear atypia; increased mitotic activity
4
Glioblastoma
Vascular proliferation or necrosis; crowded anaplastic cells; marked nuclear atypia; brisk mitotic activity
From Louis DN, Ohgaki H, Wiestler OD, Cavenee WK. World Health Organization Classification of Tumours: Pathology and Genetics of Tumors of the Nervous System. Geneva: IARC Press, 2007, with permission.
Primary or metastatic: These cells are fibrillar. They are plump and large but not spindled or vacuolated. The neoplastic cell nuclei are moderately pleomorphic; the cells have small processes emanating from them. The lesion lacks rosettes. There is a vessel cuffed by many small, reactive-appearing mononuclear cells. No epithelial elements are recognized. The findings are those of a primary CNS neoplasm rather than a metastatic lesion (Tables 10.10 and 10.11).
Glial, neuronal, and other primary considerations: Possible primary CNS neoplasm diagnoses in Table 10.10 include giant cell astrocytoma, gemistocytic astrocytoma, ganglion cell tumor, or histiocytosis (Fig. 10.1). Although nuclei of the lesional cells have a slight resemblance to neuronal nuclei, their chromatin is more condensed. Their cytoplasm is pink without purple Nissl substance of neurons. Bielschowsky stain shows no silver staining of neoplastic cells (Fig. 10.1); neoplastic cells were also negative for synaptophysin, a neuronal marker (not shown). Aggregate features do not support a ganglion cell tumor. The neoplastic cells are positive for the intermediate filament glial fibrillary acidic protein (GFAP), a marker that accentuates their fibrillarity and confirms their astrocytic nature (Fig. 10.1). The related text in this chapter describes differences between gemistocytic astrocytoma and giant cell astrocytoma. Gemistocytic astrocytomas resemble reactive astrocytes and have abundant, plump, hyaline cytoplasm and peripheralized nuclei. Macrophages (histiocytes) are GFAP-negative and lack the fibrillary structure highlighted by GFAP, arguing against the diagnosis histiocytosis. Neoplastic cells are negative for the T-lymphocyte marker CD45RO (Fig. 10.1D) and the B-lymphocyte marker CD20 (Fig. 10.1E), whereas polyclonal perivascular lymphocytes are positive. Therefore, the aggregate histologic, histochemical, and immunohistochemistry (IHC) findings support the diagnosis of gemistocytic astrocytoma.
TABLE 10.9 World Health Organization Criteria for Grading of Oligodendrogliomas
Grade
Nomenclature
Histologic Features
2
Oligodendroglioma
Moderate cellularity; homogeneously round nuclei, “fried egg” halo (paraffin); fine capillary network; mineralization (microcalcifications)
3
Anaplastic oligodendroglioma
Increased cellularity; high mitotic rate; marked cytologic atypia; microvascular proliferation; necrosis
From Louis DN, Ohgaki H, Wiestler OD, Cavenee WK. World Health Organization Classification of Tumours: Pathology and Genetics of Tumors of the Nervous System. Geneva: IARC Press, 2007, with permission.
Grade of astrocytoma: Bielschowsky stain reveals passing axons of brain tissue infiltrated by this neoplasm (Fig. 10.1). Infiltration of brain tissue is seen in astrocytomas of grade II to IV. Grade III astrocytomas have mitotic activity, but there is no evidence of mitotic activity. Features of grade IV, microvascular proliferation and necrosis, are absent. This astrocytoma is grade II. Most gemistocytic astrocytomas are grade II, although they tend to progress to a higher grade. Grading is further discussed in the “Gemistocytic Astrocytoma,” “Diffuse Astrocytoma,” and “Anaplastic Astrocytoma” sections. Other algorithmic approaches to the brain biopsy are available (9,10).
Knowledge about past medical history (e.g., previous CNS or primary neoplasms, connective tissue disease, immunosuppressive disease) is critical to interpretation. Likewise, knowledge about preoperative therapy (e.g., corticosteroids, chemotherapy, radiation therapy, radiosurgery) is also critical to interpretation of findings (e.g., necrosis, vascular fibrosis).
TABLE 10.10 Differential Diagnosis of a Mass of Fibrillar Cells | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
TABLE 10.11 Differential Diagnosis of a Mass of Epithelioid Cells | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
viewing a gross specimen. The neurosurgical lesions summarized in Tables 10.610.7,10.8,10.9,10.10,10.11,10.12,10.13,10.14,10.15,10.16,10.17,10.18 and 10.19 are usually focal, whereas the lesions in the biopsies directed toward a neurologic disease tend to be more diffuse (Table 10.2). One particularly problematic diagnosis is vasculitis, which can be focal, multifocal, or diffuse. If you are lucky enough to be consulted on any multifocal case, advise the surgeon to target a new or subacute radiographic lesion.
TABLE 10.12 Differential Diagnosis of a Mass of Conspicuously Different Cells | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
of everyone’s time. It is a ritual that helps the clinician say that everything was tried on a moribund patient.
TABLE 10.13 Differential Diagnosis of a Mass of Small Blue Cell Tumor | ||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||
TABLE 10.14 Differential Diagnosis of a Mass That Includes Syncytial Cells | ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||
TABLE 10.15 World Health Organization Criteria for Grading of Meningiomasa | ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||
TABLE 10.16 Histiocytoses Affecting the Central Nervous System Compared with Macrophages | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||
TABLE 10.17 Carcinomas and Melanoma Metastatic to Brain | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
TABLE 10.18 Differential Diagnosis of Cyst with Wall of Fibrillar Cells | ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||
TABLE 10.19 Differential Diagnosis of a Cyst with Wall Lined by Epithelium | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||
definitively and grade every case but, rather, (a) to ensure that lesional tissue has been obtained for subsequent diagnosis and grading, (b) to ensure the lesional tissue has been appropriately sampled (e.g., that high-grade features are noted on a suspected glioblastoma based on imaging features), (c) to provide sufficient preliminary diagnostic information to optimize surgery, and (d) to perform appropriate special tissue processing (Table 10.1). Optimizing surgery depends on the individual case and on whether open biopsy or stereotactic needle biopsy is being done (see later sections).
TABLE 10.20 Inclusion Immunophenotype | ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||
TABLE 10.21 Findings in Surgically Resected Temporal Lobes in Setting of Seizure Disorder | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||
History: Patient age, duration of symptoms, and surgical location are the minimum.
Radiography: View images and/or read reports. On reports, objective findings trump interpretation. Are there one or more masses? Infiltrative? Enhancing?
Check for prior specimens: Slides and reports.
Cell type: Fibrillar, epithelioid, mixed, small and crowded, or syncytial (e.g., fibrillar*)
Major features: Such as mass; marked cellular and nuclear pleomorphism*
Differential (Dif): Such as glioblastoma (GbM), giant cell astrocytoma (GCA), pleomorphic xanthoastrocytoma (PXA), sarcoma (Src), or melanoma (Mel)*
Location: Entirely extramedullary (e.g., outside of CNS [excludes GbM, GCA]), skull invasion (favors Src, Mel), subependymal (favors GCA)
Radiography: Diffuse margin with CNS (favors GbM)
Duration of symptoms: For a new mass, less than 3 months favors GbM, Src, Mel
Microscopic features:
No mitoses (favors GCA, PXA)
Granular bodies (favors PXA)
Necrosis (favors GbM, Src, Mel)
Microvascular proliferation (favors GbM)
Intercellular collagen (favors PXA, Src)
a diagnosis or monitor consequences of therapy. Biopsies can be performed using CT- or MRI-guided stereotactic needle biopsy or using an open technique. Therapeutic resections attempt gross total excision of lesional tissue. Individual cases may combine more than one procedure at a single operation. For example, open biopsy for diagnosis of an intramedullary spinal cord tumor may proceed directly to a therapeutic resection if the intraoperative evaluation suggests ependymoma.
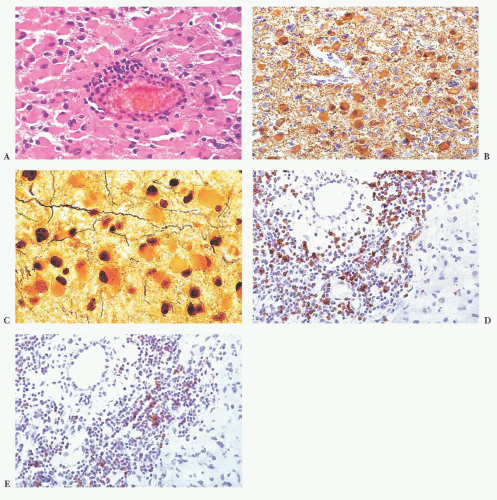 FIGURE 10.1 A solitary mass from the right frontotemporal cerebral cortex of a 38-year-old woman. This case and the first paragraphs of the text illustrate how the tables and text assist in the interpretation from the initial impression to the final diagnosis. (A) Hematoxylin and eosin stain. (B) Anti-glial fibrillary acidic protein with hematoxylin counterstain. (C) Bielschowsky silver stain for axons and neurofilaments. (D) CD45RO anti-T lymphocyte with hematoxylin counterstain. (E) CD20 anti-B lymphocyte with hematoxylin counterstain. |
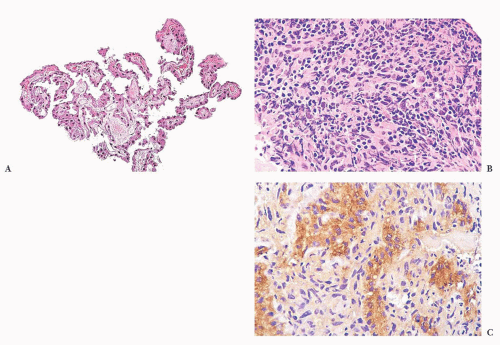 FIGURE 10.2 Stereotactic biopsy of pineal tumor. The first biopsy from this patient revealed epithelium on a fibrous base resembling choroid plexus epithelium (A). Another biopsy revealed germinoma (B). Still another specimen from this second biopsy was α-fetoprotein-positive (C), and it contained Schiller-Duval bodies (not shown) in a focus of yolk sac tumor. |
of tissue, with a comment about the need for neuroanatomical and clinical correlation to rule out the possibility of normal structure. Stereotactic biopsies of pineal tumors present problems analogous to the problems the fabled elephant presented to the six blind men (Fig. 10.2).
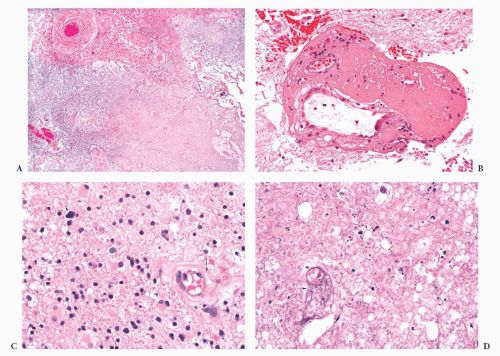 FIGURE 10.3 Treatment effects. Largely necrotic lesion in a young man a year after radiation therapy to a total dose of 81 Gy along with concurrent temozolomide chemotherapy for his glioblastoma shows (A) radiation effects, including fibrinoid necrosis of vascular walls and extensive tissue coagulation necrosis with beginning cavitation; (B) vessel has cavitation, macrophages, and fibrinoid material (FM) in its wall. Small vessels within the FM resemble revascularization in a thrombus. (C,D). Recently treated glioblastoma. One month after the end of 2 months of radiation and chemotherapy, there are (C) regions with a few remaining glioma cells (short arrow), macrophages with bubbly cytoplasmic lipids, gliosis, and attenuated vascular wall (long arrow); (D) necrosis with tiny flecks of calcium and nuclear debris; and remnants of abnormal vascular walls (arrow). This case was interpreted as treatment effects more prominent than glioma cells. (continues) |
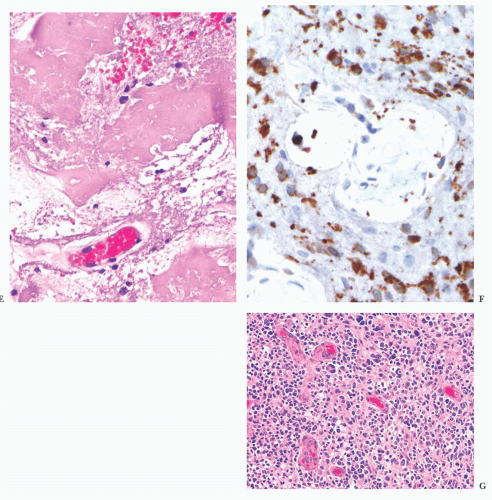 FIGURE 10.3 (Continued) Chronic radiation necrosis: (E) This man received radiation to his right temporal lobe. Years later, fibrinoid extravasation from a vessel and brain necrosis are evident. (F) Myelin basic protein from damaged white matter accumulated in macrophages. (G) Tumor progression: The original oligodendroglioma of this man was grade II. Years after radiation and chemotherapy, high cell density and numerous mitoses plus MVP lacking fibrinoid change show progression to anaplastic oligodendroglioma, grade III. |
walls. Radiation necrosis is not pseudopalisading, and during its maturation, it shows tiny flecks of calcification. Both the tumor and white matter are particularly sensitive to radiation, grey matter being more resistant. Mitoses and tumor cell density are decreased with TE.
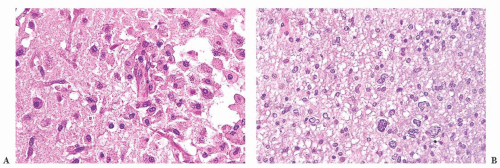 FIGURE 10.4 This frontal lobe specimen resected 24 days after an initial biopsy simulates an organizing infarct (A). The original biopsy revealed an obvious glioma (B). |
Squash/smear/touch preparations
Unfrozen routine permanent paraffin sections
Frozen sections
Electron microscopy (EM) (retain a small portion in a fixative for EM, if necessary)
Frozen tissue, for possible future molecular diagnostic studies (freeze fresh tissue as soon as possible and store)
Other (microbiology, flow cytometry, cytogenetics, molecular diagnostics)
Cassettes should be labeled with pencil as formic acid will dissolve away even the ink specially formulated for histologic processing.
Normal plastic cassettes are resistant to formic acid, but the thin plastic netting of cassettes designed for small biopsies will dissolve in formic acid, so a small biopsy should be wrapped in lens paper to keep it in the cassette. Metal tops should not be used.
features are emphasized in the following sections, under each entity. Depending on difficulty, the differential can be solved on H&E or IHC (18).
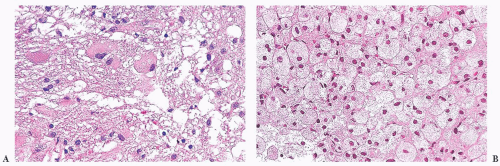 FIGURE 10.5 Reactive changes. Common reactions to injury in the central nervous system include gliosis (A) and “gitter cell” reaction of macrophages filled with neutral fat (B). The gliosis occurred in edematous brain near a metastatic carcinoma. The macrophages are in a subacute infarct clinically suspected to be a glioma. |
inflammatory, necrotic, traumatic, degenerative, and neoplastic processes (21,22 and 23). The origin of these cells is variable among lesions and consists of some combination of (a) “native” parenchymal microglial cells, which can proliferate along with (b) blood-derived macrophages entering the brain parenchyma in response to damage (22). The fate of the latter cells may be to remain at the site of injury or migrate to the perivascular space (23). Because distinction among these origins is not useful diagnostically, the inclusive term macrophages can be employed.
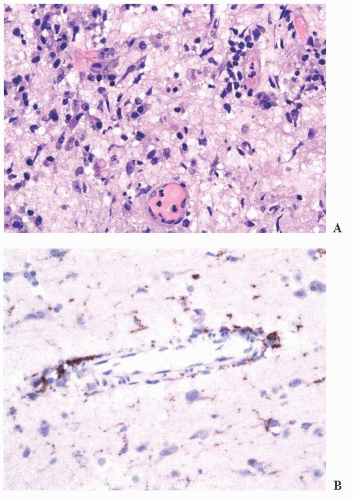 FIGURE 10.6 (A) Perivascular mixed inflammatory infiltrate is composed of lymphocytes, plasma cells, and macrophages around but not within vessel walls. (B) Chronic encephalitis stained with CD68 shows elongated microglial macrophages within brain tissue and around vessels but not within the vascular wall. Note the normal thin endothelial lining (compare with vasculitis, Fig. 10.25C,D). |
wounds and implants (24). Macrophages ingest the irritant or injured cellular constituents and move them to the perivascular space (23). In the absence of classic lymph nodes in the brain, this perivascular region is where cells that respond to antigens intermingle. Depending on severity and duration of illness, the perivascular inflammation varies substantially. Old hemorrhage is characterized mainly by perivascular macrophages laden with hemosiderin. Viral or autoimmune encephalitis produces a maximal response, with abundant perivascular macrophages and lymphocytes along with parenchymal infiltrates of microglial cells and microglial nodules.
modalities of variable severity. The etiology is often elusive, but etiologies include infectious (herpes simplex virus [HSV], CMV, etc.), inflammatory (multiple sclerosis [MS], connective tissue disease, etc.), vascular (infarct due to thrombosis), developmental (vascular malformation), traumatic, and iatrogenic processes (29,30). Workup is based on imaging and CSF and serum evaluation; biopsy is occasionally performed to search for an etiology.
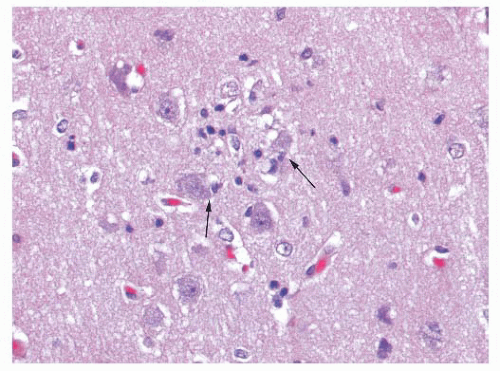 FIGURE 10.7 Chronic encephalitis. The hallmark of chronic encephalitis is neuronophagia, the “eating of neurons.” Lymphocytes recognize infected neurons, even ones such as these that show no inclusions, and direct microglial macrophages to eat them (arrows). Notice that these infected neurons are not hypoxic; they have not lost their purple cytoplasmic Nissl substance nor have they turned orange. When there is no encephalitis, the most common cause of neuronophagia is hypoxic damage to neurons. |
(e.g., from a primary pulmonary source) or via direct extension from an infected sinus.
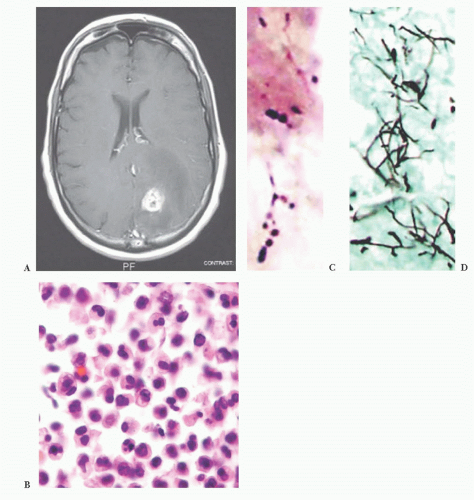 FIGURE 10.8 Abscess in a 49-year-old man who had a first seizure after a dental procedure. (A) Imaging revealed an enhancing cystic lesion. (B) Suppurative inflammation was noted, including neutrophils with admixed macrophages and lymphocytes. (C) Gram stain demonstrates filamentous bacteria. (D) Grocott methenamine silver stain of filamentous bacteria. Actinomycosis was cultured. |
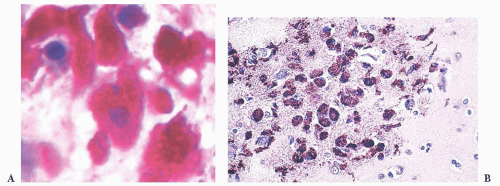 FIGURE 10.9 Whipple disease of the central nervous system. Typical cluster of reactive cells stained with periodic acid-Schiff stain (A) and by immunoperoxidase for anti-group B Streptococcus (B). (B, Courtesy of Dr. M. E. Velasco, Medical College of Ohio, Toledo, OH.) |
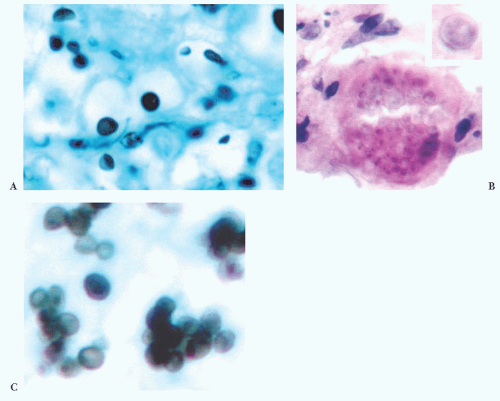 FIGURE 10.10 (A) Cryptococcosis in the subarachnoid space with a capsule (Grocott methenamine silver). (B) Coccidioidomycosis spherule and free endospore (inset) from a necrotizing brain lesion (hematoxylin and eosin). (C) Histoplasmosis in macrophages and free in brain tissue in a necrotizing brain lesion (Grocott methenamine silver). |
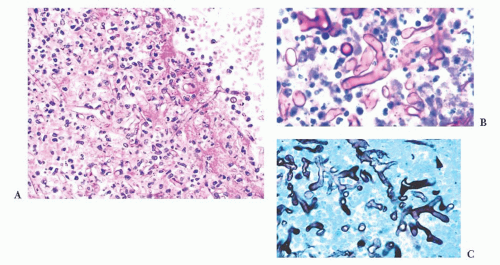 FIGURE 10.11 Zygomycetes. (A) Vessel wall is diffusely invaded by hyphal organisms with associated chronic inflammatory cell infiltrate. (B) Periodic acid-Schiff and (C) Grocott methenamine silver stains highlight the fungal morphology. |
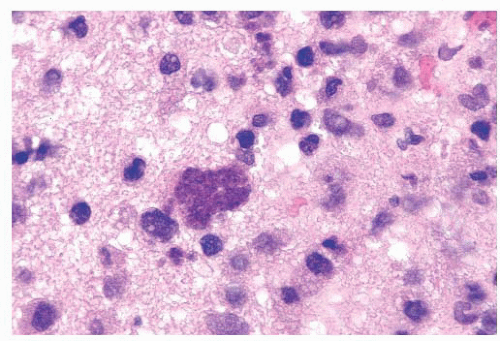 FIGURE 10.12 Toxoplasmosis in an immunodeficient patient. Amphophilic pseudocyst full of a cluster of bradyzoites is irregular and larger than surrounding cells. |
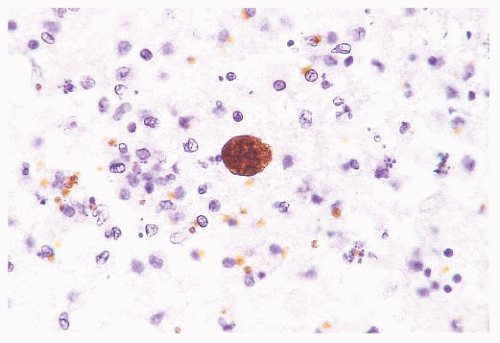 FIGURE 10.13 Cyst and free tachyzoites immunostained for Toxoplasma antigen (immunoperoxidase with hematoxylin counterstain). |
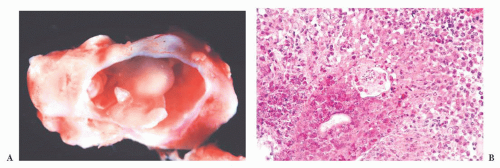 FIGURE 10.14 (A) Cysticercosis cyst resected from the cerebral hemisphere of a young South American man with seizures. (B) Degenerating schistosome egg at the center surrounded by heavy mixed inflammatory infiltrate. (A, Courtesy of Dr. D. Burns, University of Texas Southwestern, Dallas, TX.) |
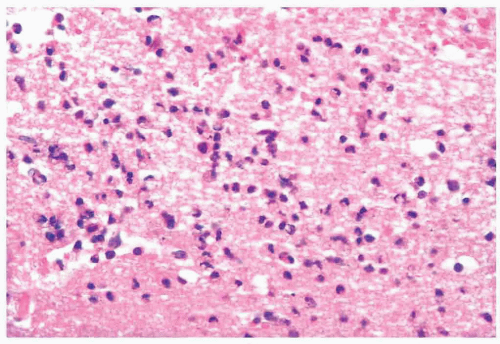 FIGURE 10.15 Inflammatory infiltrate in herpes simplex virus encephalitis. (Courtesy of Dr. F. Stephen Vogel, Duke University Medical Center, Durham, NC.) |
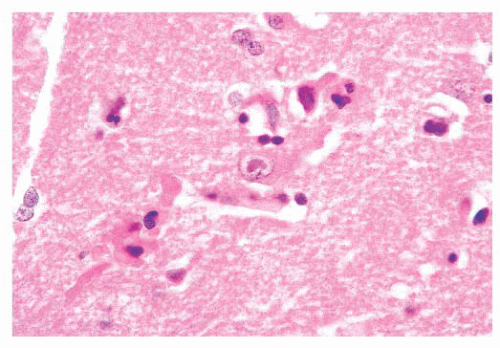 FIGURE 10.16 Herpes simplex virus encephalitis. Cowdry type A inclusion body at the center. (Courtesy of Dr. F. Stephen Vogel, Duke University Medical Center, Durham, NC.) |
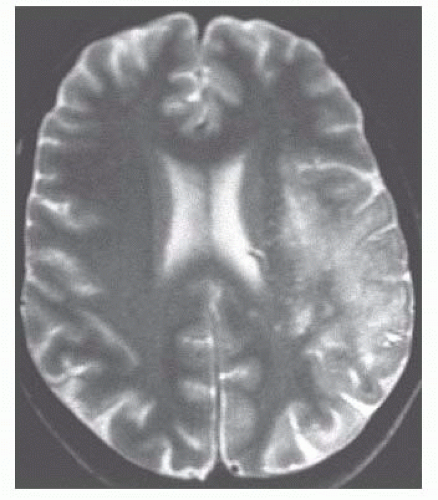 FIGURE 10.17 Progressive multifocal leukoencephalopathy simulates an edematous mass in this T2-weighted magnetic resonance image from a patient with severe immunodeficiency. (Courtesy of Dr. Herbert Ichinose and Dr. Joseph Epps, Jr., Pendleton Memorial Methodist Hospital, New Orleans, LA.) |
IgG and IgM in both the CSF and serum. Brain biopsy is rarely performed. When undertaken, viral encephalitis-type changes predominate and there is loss of myelin in subcortical white matter (28). Nuclear inclusions may be detected in neurons and oligodendroglial cells by H&E. IHC or ISH evaluation detects measles virus antigen.
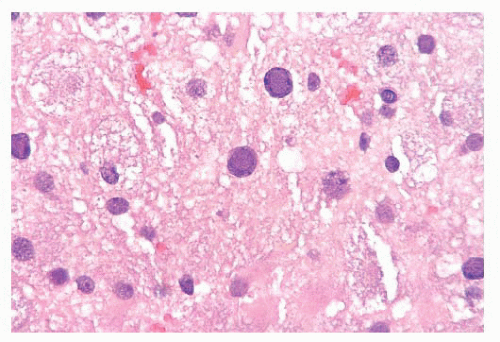 FIGURE 10.18 Progressive multifocal leukoencephalopathy. An infected oligodendroglial nucleus is enlarged by a pink-purple (amphophilic) accumulation of virions and peripheralization of chromatin. |
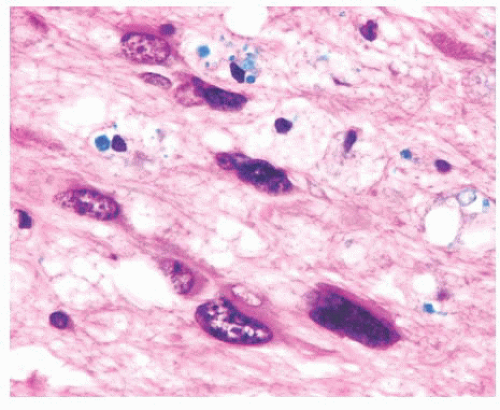 FIGURE 10.19 Deep white matter lesion from a 54-year-old woman treated with immunosuppressives for lupus. Astrocytes manifest strikingly atypical nuclei. Abundant foamy macrophages contain blue balls of myelin, which is consistent with demyelination. |
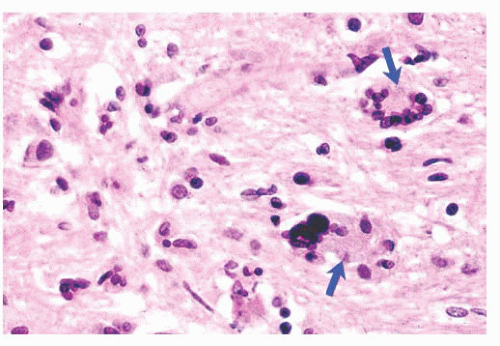 FIGURE 10.20 Human immunodeficiency virus encephalitis. Multinucleated giant cells (arrows) in the setting of a loosely formed microglial nodule accompanied by gliosis. (Courtesy of Dr. Javad Towfighi, Penn State University, Hershey, PA.) |
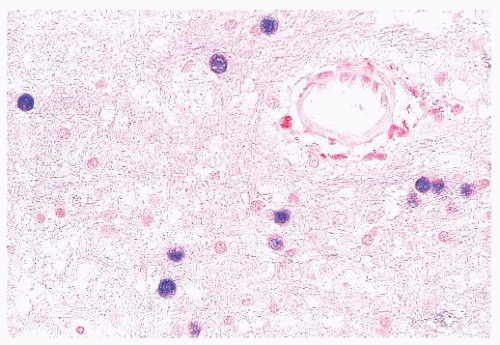 FIGURE 10.21 Progressive multifocal leukoencephalopathy. Cytochemical detection of JC viral sequences by in situ hybridization in larger nuclei stained blue by alkaline phosphatase reaction product. Negative nuclei counterstain pink, and perivascular macrophages contain light brown cytoplasmic granules. (Courtesy of Dr. Ricardo Lloyd, Mayo Clinic, Rochester, MN.) |
produce vacuoles that encircle neurons, vessels, and glia; these are larger than CJD vacuoles. Nonetheless, because many vacuoles are not related to prions, molecular diagnosis of prions is needed to confirm suspicions based on vacuoles.
 FIGURE 10.22 Creutzfeldt-Jakob disease. Biopsy of the cerebral frontal lobe from a 57-year-old man with suspected Alzheimer disease. Coalescing round vacuoles and gliosis are present in the neuropil. Some of the vacuoles have a glassy appearance rather than a punched out look. These involved all six cortical layers (cresyl violet with eosin). |
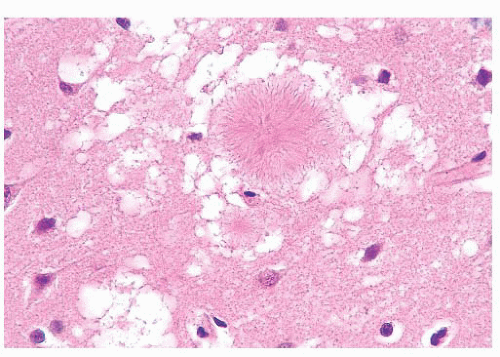 FIGURE 10.23 Variant Creutzfeldt-Jakob disease. This variant is from the cerebral cortex of a 29-year-old woman in the United Kingdom who experienced 17 months of progressive dementia, ataxia, and chorea. Fibrillary plaques composed of prion protein resemble skyrocket bursts, and they are surrounded by very coarse spongiform changes in the neuropil. (Courtesy of Dr. Jeanne E. Bell, National CJD Surveillance Unit, Western General Hospital, Edinburgh, United Kingdom.) |
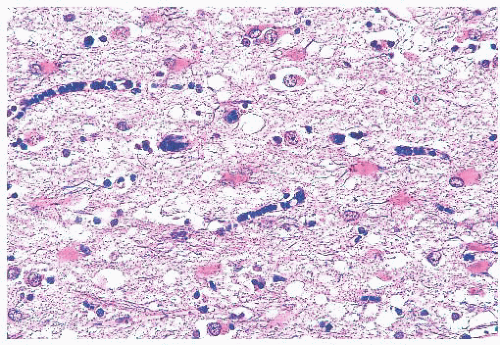 FIGURE 10.24 Familial Creutzfeldt-Jakob disease. Most of the neurons have been lost and replaced by gliosis in this case. Phosphotungstic acid hematoxylin stain highlights gliotic fibrils and vacuolated neuropils. |
fraction of axons are lost in chronic MS. In contrast, axonal loss precedes myelin loss in an infarct (Fig. 10.26). The amount of gliosis increases with the chronicity of the lesion.
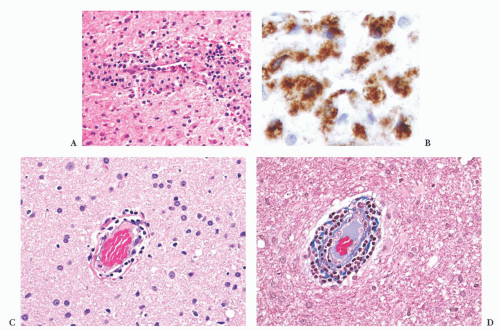 FIGURE 10.25 Vascular involvement in multiple sclerosis (A,B) compared with vasculitis (C,D). Active MS lesion presented on MRI as ring-enhancing lesion in a 24-year-old woman. (A) Abundant plump cells and scant lymphocytes around blood vessel but not in its wall. Initial impression could mistake this lesion for vasculitis or glioma. (B) CD68 immunostain identifies the plump cells as macrophages. Active vasculitis presented on MRI as severe white matter disease with patchy enhancement in a 32-year-old woman. (C) Lymphocytes and macrophages penetrate the pink necrotic wall of the vessel and insinuate between collagen fibers. Endothelium is swollen. (D) Trichrome stain reveals vascular wall penetration and plump endothelium. |
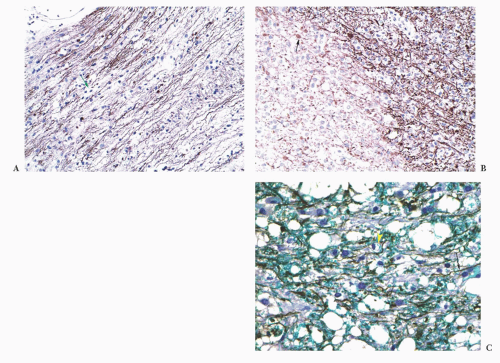 FIGURE 10.26 Two different white matter lesions (A,B) are each stained for neurofilaments, with hematoxylin counterstain for nuclei. (A) Demyelination is hard to see except for some pale, lipid-filled macrophages (arrow) between straight brown axons. Axons are spared in demyelination. (B) Focal white matter necrosis produced the pale side of this image in this small infarct. Necrotic axons become very pale and finally disintegrate into small beads that are eaten by macrophages. Some damaged axons swell (arrow). (C) Vacuolar myelopathy as seen in vitamin deficiency, metabolic disorders, toxins, and HIV where vacuolation and demyelation with naked axons (arrow) and blue balls of myelin, some in macrophages (arrowhead), precede axonal damage. Neurofilaments/Luxol fast blue/hematoxylin. |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


