Ewing Sarcoma/PNET of Soft Tissue
Cyril Fisher, MD, DSc, FRCPath
Key Facts
Terminology
Family of small round cell translocation-associated sarcomas occurring in bone, soft tissue, or skin
Additionally, PNET shows neural differentiation
Clinical Issues
Mostly children and young adults
Painful or painless mass
5-year survival: 65-90%
25% in those presenting with metastatic disease
Microscopic Pathology
Sheets of closely packed uniform cells
Round uniform nuclei
Scanty cytoplasm, sometimes clear
Homer-Wright rosette formation in PNET
Atypical (large cell) variant
Conspicuous nucleoli
Ancillary Tests
PAS stains glycogen in cytoplasm
Several translocations involving EWSR1 and ETS family genes
Most common are EWSR1-FLI1 (90%) and EWSR1-ERG (8-10%)
Top Differential Diagnoses
Alveolar rhabdomyosarcoma
Poorly differentiated synovial sarcoma
Desmoplastic small round cell tumor
Neuroblastoma
Small cell carcinoma
Extraskeletal mesenchymal chondrosarcoma
Lymphoma
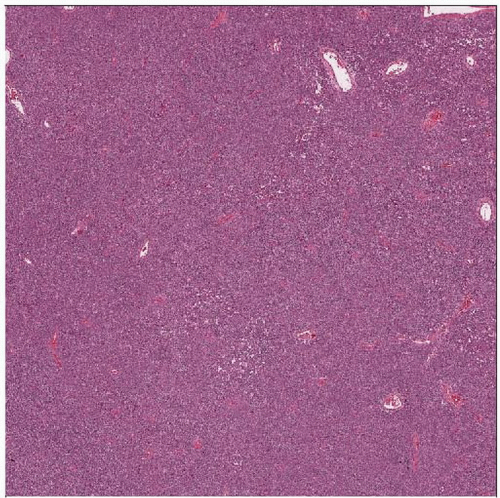 Ewing sarcoma and PNET are usually extremely cellular, with a dense, solid to sheet-like distribution of cells. A delicate vascularity is noted, but overall this is a solid population of cells. |
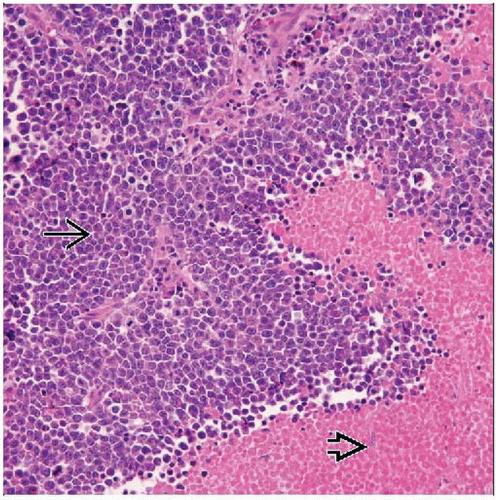 This is a very cellular tumor arranged in a diffuse sheetlike pattern  . Geographic areas of coagulative necrosis are easily identified . Geographic areas of coagulative necrosis are easily identified  . There is often perivascular viable tumor. . There is often perivascular viable tumor. |
TERMINOLOGY
Abbreviations
Ewing sarcoma (ES)
Primitive neuroectodermal tumor (PNET)
Definitions
Family of small round cell translocation-associated sarcomas occurring in bone or soft tissue
Extraskeletal ES
Small cell tumor of thoracopulmonary region (Askin tumor)
PNET of soft tissue
Same morphologic spectrum as ES
Additionally shows neural differentiation
Several translocations known
Similar changes in all types and sites
CLINICAL ISSUES
Epidemiology
Incidence
Rare
Age
Mostly children and young adults
Sporadic cases at any age
Gender
Slight male predominance
Ethnicity
More common in Caucasians
Very rare in Africans and African-Americans
Site
Lung, mediastinum, paravertebral region
Retroperitoneum, abdominal cavity
Limbs (deep soft tissue), skin (rare)
Primary bone tumor can extend and present in soft tissue
Rarely in viscera (kidney, uterus, pancreas, larynx)
Presentation
Painful or painless mass
Systemic symptoms
Fever, anemia, leukocytosis
Treatment
Options, risks, complications
Chemotherapy is treatment of choice
Surgical excision of residual masses
Irradiation in selected cases
Drugs
Multiagent
At least 3 of vincristine, doxorubicin, cyclophosphamide, ifosfamide, etoposide
Potential biologic therapies
Anti-CD99 antibodies
Small interfering RNA against EWSR1-FLI1 gene product
Trastuzumab (against EGFR2), figitumumab (against IGF-1R)
Prognosis
5-year survival: 65-90%
25% in those presenting with metastatic disease
Locally infiltrative
Metastasizes to bone, lungs
Presence of EWSR1-FLI1 rearrangement is prognostically favorable
Cutaneous and subcutaneous tumors have improved prognosis
MACROSCOPIC FEATURES
General Features
Pale friable tumor
Hemorrhage and necrosis
Can be cystic
Size
Variable, up to 20 cm
MICROSCOPIC PATHOLOGY
Histologic Features
Sheets of closely packed uniform cells
Sometimes divided into lobules
Pseudoalveolar pattern
Round uniform nuclei
Dispersed fine chromatin, inconspicuous nucleoli
Scanty cytoplasm
Can be clear due to glycogen
Cells rarely spindled focally
Homer-Wright rosette formation in PNET
Central fibrillary core without lumen
Minimal intercellular collagen or reticulin
Rare myxoid change
Stromal microcysts sometimes present
Necrosis frequent
Perivascular preservation
Rare morphologic variants
Atypical (large cell) variant
Larger, vesicular nuclei
Irregular nuclear outline
Conspicuous nucleoli
Pleomorphism, spindling
Adamantinoma-like variant occurs mostly in bone
Sclerosing and clear cell variants described
ANCILLARY TESTS
Histochemistry
PAS
Reactivity: Positive for glycogen
Staining pattern
Cytoplasmic
Staining is abolished by pretreatment with diastase
Reticulin
Reactivity: Negative
Staining pattern
Minimal or no intercellular reticulin except around blood vessels
In Situ Hybridization
FISH with break-apart probe for EWSR1 gene (at 22q12) shows rearrangement
Molecular Genetics
Several balanced translocations and fusions involving EWSR1 and ETS family genes
t(11;22)(q24;q12), EWSR1-FLI fusion (90% of cases)
t(21;22)(q12;q12), EWSR1-ERG fusion (5-10% of cases)
t(2;22)(q33;q12), EWSR1-FEV fusion (< 1% of cases)
t(7;22)(p22;q12), EWSR1-ETV1 (< 1% of cases)
t(17;22)(q12;q12), EWSR1-E1AF (< 1% of cases)
Rarely others in occasional cases
Genetic changes do not correlate with site or morphology
DIFFERENTIAL DIAGNOSIS
Alveolar Rhabdomyosarcoma
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


