Epilepsies with Onset at All Ages: Symptomatic and Probably Symptomatic Focal Epilepsies
One group of epilepsies has onset in patients of all ages: symptomatic and probably symptomatic focal epilepsies. These epilepsies are listed in Table 3-1.
Based on seizure and electroencephalographic (EEG) characteristics, age, and evidence of brain pathology, a patient with symptomatic focal epilepsies often can be classified into one of several epilepsy syndromes, according to the presumed site in which seizures originate: mesial temporal lobe, lateral temporal lobe, frontal lobe, parietal lobe, or occipital lobe. Rasmussen syndrome and hemiconvulsion-hemiplegia syndrome also are included in this epilepsy group.
I. TEMPORAL LOBE EPILEPSIES
A. General Characteristics
Simple partial, complex partial, or secondarily generalized seizures (reviewed in Chapter 2) may occur, with onset frequently in childhood or young adulthood. Seizures may occur randomly, at intervals, or in clusters. Simple partial seizures are characterized by autonomic or psychic symptoms, or both, and by certain sensory phenomena, such as olfactory and auditory illusions or hallucinations. The most common sensation is a rising epigastric discomfort.
B. Routine Electroencephalogram Characteristics
Routine EEGs may show (a) no abnormality; (b) slight or marked asymmetry of the background activity; or (c) temporal spikes, sharp waves, or slow waves (unilateral or bilateral, synchronous or asynchronous; may not be confined to temporal areas).
C. Subtypes
1. Mesial Temporal Seizures
Mesial temporal seizures are the most common form of temporal lobe epilepsy and generally conform to the following general description. Seizures are characterized by rising epigastric discomfort, nausea, marked autonomic signs, and other symptoms including borborygmi, belching, pallor, fullness of the face, flushing, arrest of respiration, pupil dilation, fear, panic, and olfactory-gustatory hallucinations. Scalp EEG often shows unilateral or bilateral spikes most prominent in the anterior temporal leads.
One variant of amygdala-hippocampal seizures is called the mesial temporal lobe epilepsy syndrome. Such patients demonstrate
mesial temporal sclerosis (Fig. 3-1). They typically have a strong family history of epilepsy showing an autosomal-dominant inheritance with incomplete penetrance. The patient has seizures (often complicated) during infancy or childhood. After a silent period lasting 2 to 15 years, unprovoked partial seizures begin in late childhood or early adolescence. The seizures are refractory to medical treatment in 20% to 30% of patients.
mesial temporal sclerosis (Fig. 3-1). They typically have a strong family history of epilepsy showing an autosomal-dominant inheritance with incomplete penetrance. The patient has seizures (often complicated) during infancy or childhood. After a silent period lasting 2 to 15 years, unprovoked partial seizures begin in late childhood or early adolescence. The seizures are refractory to medical treatment in 20% to 30% of patients.
Table 3-1. Groups of epilepsy syndromes and specific epilepsy syndromes with onset at all ages and accompanying seizure types | |||||||
|---|---|---|---|---|---|---|---|
|
2. Lateral Temporal Seizures
Lateral temporal seizures begin as simple partial seizures characterized by auditory hallucinations or illusions, dreamy state, visual misperceptions, or language disorders (dominant-hemisphere focus). These may progress to complex partial seizures if propagation to mesial temporal or extratemporal structures occurs. Lateral temporal seizures usually lack several of the features typical of mesial temporal seizures, including automatisms, contralateral dystonia, swerving head movements, body shifting, hyperventilation, and postictal cough or sigh. The scalp EEG often shows unilateral or bilateral spikes most prominent in the middle or posterior temporal leads.
A special subtype is termed autosomal-dominant partial epilepsy with auditory features (ADPEAF). Onset is in typically in the second decade of life. The syndrome is characterized by auditory symptoms. The auditory phenomenon may consist of unformed sounds such as monotonous buzzing or fringing, distortions of sound such as a change in volume or pitch, or formed sounds such as voices from the past or specific singers. Other sensory or psychic symptoms, motor activity, and autonomic dysfunction may develop. Paroxysmal activity may be seen in the EEG interictally in the temporal or occipital leads. The condition responds to antiepileptic drugs, but may require prolonged administration. Genetic analysis has found linkage to chromosome 10q24 with mutations in the LGI1 gene (leucine-rich gene, glioma inactivated). The gene is involved in ligand binding and protein-protein interactions and likely is involved in development of the nervous system. LGI1 is the only gene yet identified in temporal lobe epilepsy and is the only non-ion-channel gene identified in idiopathic epilepsy.
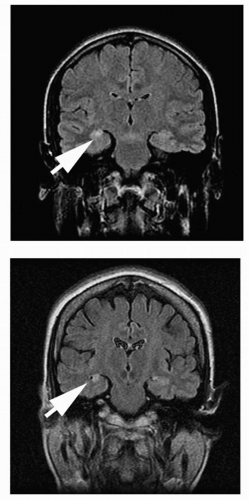 Fig. 3-1. MRI of patient with mesial temporal sclerosis. FLAIR images showing signal changes in the right mesial temporal region (arrows). |
3. Lateralizing Features
Useful lateralizing features for the temporal lobe seizures include unilateral clonic activity (seizure focus contralateral in all patients); unilateral dystonic or tonic posturing (seizure focus contralateral in 90% or 86%, respectively); unilateral automatisms (seizure focus ipsilateral in 80%); and ictal speech preservation (seizure focus contralateral to the language-dominant hemisphere in 80%). Versive head rotation occurring less than 10 seconds before seizures secondarily generalize predicts a contralateral focus. Ictal speech arrest or postictal speech impairment is associated with a seizure focus ipsilateral to the language-dominant hemisphere in two thirds of patients. Postictal speech preservation is associated with a seizure focus contralateral to the language-dominant hemisphere in two thirds of patients. Seizure manifestations not providing reliable lateralizing information include eye deviation, type of aura, and versive head movements occurring at times other than immediately before seizures secondarily generalize.
4. Cysticercosis
As mentioned in the opening paragraph of this chapter, the manifestations of partial seizures depend on the region of the cortex in which they originate. This is true for cysticercosis, a common cause of partial seizures in developing countries and certain regions of the United States in which it is endemic. The diagnosis is best established with imaging studies.
II. FRONTAL LOBE EPILEPSIES
A. Clinical Characteristics
Frontal lobe epilepsies are characterized by simple partial, complex partial, or secondarily generalized seizures (reviewed in Chapter 2), or combinations of these. Features suggesting frontal lobe epilepsies are (a) frequent seizures, often in stage 2 sleep; (b) short seizure duration; (c) minimal or no postictal confusion after a complex partial seizure; (d) rapid secondary generalization; (e) prominent motor manifestations that are tonic or postural; (f) complex gestural automatisms (may be sexual) at onset; (g) frequent falling during the seizure; and (h) frequent episodes of status epilepticus.
B. Electroencephalogram Characteristics
The interictal EEG of frontal lobe epilepsy patients may show (a) no abnormality; (b) background asymmetry; and (c) spikes, sharp waves, or paroxysmal fast activity that can be unilateral or bilateral, unilobular, or multilobular. Patients whose seizures originate from the dorsolateral convexity tend to have interictal epileptiform abnormalities that localize to the region of seizure onset. Patients whose seizures begin in the medial frontal region tend to have either no epileptiform activity or multifocal epileptiform discharges. Vertex or midline epileptiform discharges also can be seen with medial frontal foci. Frontal foci not infrequently exhibit spikes or sharp waves in the temporal leads.
C. Subtypes
1. Supplementary Motor Seizures
Supplementary motor seizures are typically brief, lasting only 10 to 40 seconds. The patient demonstrates abrupt tonic posturing of one or more extremities; the arms are affected more often than the legs. Characteristically, arms and legs are tonically adducted. During the tonic phase, the patient may cry or moan loudly. Consciousness is usually preserved, but the patient may be unable to speak. A versive movement, usually away from the side of ictal onset, may precede secondary generalization. The tonic posturing may be preceded by sensory symptoms in an extremity. Supplementary motor seizures occur frequently, and for a patient to experience five to ten episodes per day is not rare. Many seizures occur during sleep. Commonly, these seizures are medically intractable.
2. Cingulate
Cingulate seizure patterns are complex partial with complex motor gestural automatisms at onset. Autonomic signs are common, as are changes in mood and affect.
3. Anterior Frontopolar Region
Anterior frontopolar seizure patterns include forced thinking or initial loss of contact and adversive movements of head and eyes, with possible evolution, including contraversive movements and axial clonic jerks, and falls and autonomic signs.
4. Orbitofrontal
The orbitofrontal seizure pattern is one of complex partial seizures with initial motor and gestural automatisms, olfactory hallucinations and illusions, and autonomic signs. Automatisms may include unformed or formed speech (including expletives) and walking around the room.
5. Combined Mesial Frontal
Seizures originating in any of the four mesial frontal structures described earlier sometimes show phenomena described for other mesial frontal structures. Functional spread of discharges likely occurs among the areas.
6. Dorsolateral
Dorsolateral seizure patterns may be tonic or, less commonly, clonic, with versive eye and head movements and speech arrest.
7. Opercular
Opercular seizure characteristics include mastication, salivation, swallowing, laryngeal symptoms, speech arrest, epigastric aura, fear, and autonomic phenomena. Simple partial seizures, particularly partial clonic facial seizures, are common and may be ipsilateral. If secondary sensory changes occur, numbness may be a symptom, particularly in the hands. Gustatory hallucinations are particularly common with seizures in this area.
8. Motor Cortex
Motor cortex epilepsies are characterized mainly by simple partial seizures, and their localization depends on the side and topography of the area affected. In cases of the lower prerolandic area, speech arrest, vocalization or dysphasia, tonic-clonic movements of the face on the contralateral side, or swallowing may occur. Generalization of the seizure frequently occurs. In the rolandic area, partial motor seizures with march, or jacksonian, seizures occur, particularly beginning in the contralateral upper extremities. In the case of seizures involving the paracentral lobule, tonic movements of the ipsilateral foot may occur, as well as contralateral leg movements. Postictal paralysis is frequent.
9. Kojewnikow Syndrome
Kojewnikow syndrome represents a particular form of rolandic partial epilepsy both in adults and in children and is related to a variety of lesions in the motor cortex. Its principal features are (a) motor partial seizures, always well localized; (b) often late appearance of myoclonus in the same site at which somatomotor seizures occur; (c) an EEG with normal background activity and a focal paroxysmal abnormality (spikes and slow waves); (d) occurrence at any age in childhood and adulthood; (e) frequently demonstrable etiology (tumor, vascular); and (f) no progressive evolution of the syndrome (clinical, EEG, or psychological, except in relation to the evolution of the causal lesion).
10. Rasmussen Encephalitis
In Rasmussen encephalitis, in a previously normal child, usually approximately 6 to 10 years old, therapy-resistant focal seizures rapidly develop, usually motor or sensorimotor, with a slowly progressive motor deficit implicating the same cerebral hemisphere. A mild or moderate mental deficit appears later. The EEG shows prominent and persistent arrhythmic delta waves, loss of background features, and abundant spikes. Later, seizures may implicate widely separate portions of the same hemisphere. Pathologic specimens may show gliosis, inflammation, or spongiform changes. The disease may progress to death, stabilize, or improve over time. High-dose intravenous human immunoglobulin may be beneficial for this condition.
11. Autosomal-Dominant Frontal Lobe Nocturnal Epilepsy
Autosomal-dominant frontal lobe nocturnal epilepsy is a syndrome in which nocturnal seizures develop within the first two decades of life, although the seizures often persist throughout adulthood. The seizures usually occur during sleep, but in severe cases, seizures may occur while awake. The clinical features are similar to other frontal epilepsies. A nonspecific aura that may include somatosensory, special sensory, psychic, or autonomic phenomena is commonly present. After the aura, gasping, groaning, other vocalization, prominent motor phenomena (thrashing, hyperkinetic movements, tonic stiffening, clonic jerking), or reflex agitation with rapid changes in position may occur. The condition usually responds well to carbamazepine.
The gene defect has been mapped to chromosome 20q13 (CHRNA4) and 1q21 (CHRNB2) with mutations in the genes coding for the α4 and β2




Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
