I. MECHANISM OF ACTION
Marketed antiepileptic drugs work by varying combinations of mechanisms.
A. Sodium Currents
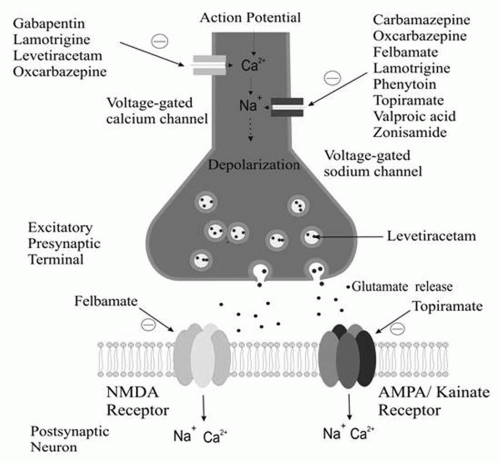
The firing of an action potential by an axon requires passage of sodium into the axon through sodium channels. Sodium channels exist in three states: (a) resting (able to allow passage of sodium), (b) active (allowing passage of sodium), and (c) inactive (not allowing passage of sodium). After activation, a percentage of sodium channels become inactivated for a period of time. With repetitive axonal firing, enough sodium channels become inactivated that the axon can no longer propagate an action potential. Some antiepileptic drugs stabilize the inactive form of sodium channels, preventing its return to the active state. This, in turn, prevents sustained repetitive firing of the axon. Drugs such as phenytoin and carbamazepine, felbamate, lamotrigine, topiramate, oxcarbazepine, valproate, and zonisamide have been demonstrated to attenuate voltage-gated Na+ channels in a usedependent fashion.
B. γ-Aminobutyric Acid (GABA)A Receptor Currents
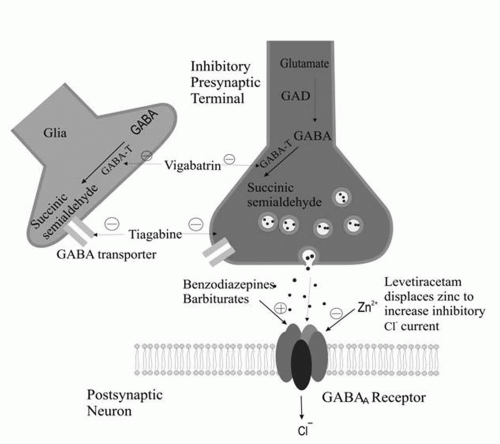
Attachment of γ-aminobutyric acid (GABA) to GABAA receptors facilitates the passage of chloride ions through chloride channels into cells. Chloride ions are negatively charged. Their passage into the cell makes the resting membrane potential more negative inside the cell and makes it more difficult for the cell to depolarize. Some antiepileptic drugs are agonists of this type of GABA-mediated chloride conductance. The GABAA receptor has benzodiazepine and barbiturate receptor sites. Activation of the benzodiazepine receptor enhances the frequency of openings of the GABAA receptor. Activation of the barbiturate receptor increases the duration of openings of the GABAA receptor.
A large number of AEDs exert their effects through enhanced inhibition by augmenting the effect of GABA. This is accomplished by the drug acting directly at the GABAA site, allosterically influencing the chloride current (barbiturates, benzodiazepines, and felbamate), by antagonizing neuronal and glial reuptake of GABA (tiagabine), or by interfering with the metabolic breakdown of GABA (vigabatrin). Valproate and gabapentin increase GABA synthesis and turnover.
C. Reduction of Voltage-sensitive Calcium Currents
Calcium ions are positively charged. Their passage into a cell renders the cell membrane less polarized. T-calcium currents are low-voltage, rapidly inactivated currents that act as pacemakers for normal rhythmic brain activity, especially in the thalamus. T-calcium currents also appear to have an important role in pacing the three-per-second spike-wave activity of absence seizures (but not other seizure types). Drugs inhibiting T-calcium currents specifically inhibit absence seizures.
The L-, N-, P-, R-, and Q-calcium channels are high-voltage activated channels distinct from low-voltage T-calcium currents. Levetiracetam, lamotrigine, phenobarbital, felbamate, and topiramate target high-voltage calcium channels. Gabapentin and pregabalin bind to the α2δ subunit of the P/Q voltage-gated calcium channel. As a result, gabapentin and pregabalin reduce the calcium current at the terminal, reducing the release of glutamate, noradrenaline, and substance P.
D. Glutamate-receptor Antagonists
Excitation in the human nervous system is produced principally by binding of the excitatory amino acid glutamate to three types of ionotropic glutamate receptors: N-methyl-D-aspartate (NMDA), α-amino-3-hydroxy-5-methyl-4-isoxazole propionate (AMPA), and kainate. Binding of glutamate to these receptors facilitates passage of calcium and sodium into the cell and potassium out of the cell. The net effect is reduction of the negative resting-membrane potential, making the cell less electrically stable. Some antiepileptic drugs act by antagonism of one or more types of glutamate receptor. Felbamate has a dual action, blocking NMDA channels and enhancing the GABAergic effect. Topiramate has been shown to attenuate non-NMDA (AMPA) and kainate currents.
E. Other Mechanisms
Recently, another target of AEDs, the H (hyperpolarization induced, cyclic nucleotide gated) channel, has been characterized. The H-channel has a high permeability to potassium and tends to stabilize the membrane potential toward the resting potential against both hyperpolarizing and depolarizing inputs. Drugs such as lamotrigine and gabapentin target the H-channels by increasing the hyperpolarization-activated cation current.
The mechanism of action of levetiracetam is not entirely clear. However, the drug blocks N-type Ca2+ channels and reverses the inhibition by the negative allosteric modulators zinc and β-carbolines on neuronal GABA- and glycine-gated currents. In addition, the drug binds to a synaptic protein (SV2). How binding to a synaptic protein results in reduced excitability (or enhanced inhibition) is not clear.
Figures 11-1 and
11-2 depict the primary mechanisms of antiepileptic drug action at excitatory and inhibitory synapses.
III. BASIC PHARMACOKINETIC PRINCIPLES
An understanding of basic principles of pharmacokinetics and pharmacodynamics of antiepileptic drug therapy is necessary for the optimal management of patients with epilepsy.
Pharmacokinetics is a measure of the action of drugs in the body over a period of time, including the processes of absorption, distribution, localization in tissues, biotransformation, and excretion. Pharmacodynamics is the study of the biochemical and physiological effects of drugs and the mechanisms of their actions, including the correlation of action and effects of drugs with their chemical structure. Whereas pharmacokinetic data are critical in determining dosage, frequency of administration, and time of steady state, pharmacodynamics provides information about efficacy and side effects of the administered drugs. In other words, pharmacokinetics is the study of what the body does to a drug, whereas pharmacodynamics is the study of what a drug does to the body.
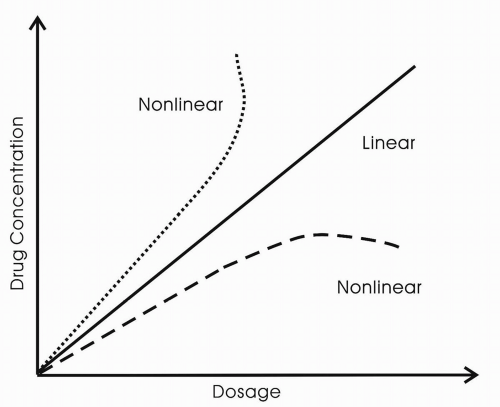
The drug’s bioavailability provides information about the degree to which a drug becomes available to the target tissue after administration. Bioavailability is dependent on absorption and first-pass hepatic metabolism, in the case of oral administration. In general, most antiepileptic drugs are well absorbed. The one exception is gabapentin, for which the rate of absorption is dose dependent. The drug is transported into the blood from the gut by a saturable transport mechanism, the L-amino-acid transport system. Because of this dose-dependent bioavailability, plasma concentrations of the drug are not directly proportional to dose throughout the range of doses administered. With increasing dosages of gabapentin, no proportional increase in blood level occurs.
Compared with adults, drug absorption is compromised in newborns but is increased in children. Food does slow the rate of absorption (but not total amount absorbed), and rapid transit through the gut can reduce blood levels. In most cases, it is not important whether the drug is taken with or without food. However, it is recommended that the patients be consistent and take the drug either with or without food on a regular basis. Because rapid absorption of tiagabine can result in toxicity, it is recommended that tiagabine be taken with food.
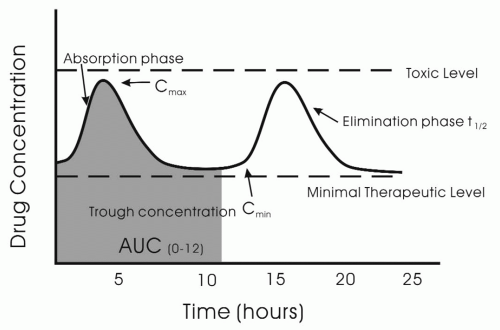
The peak concentration of the antiepileptic drug, C
max, is related to the rapidity of absorption (
Fig. 11-3). The C
max is often is the time when the drug should have its greatest effect and also is most likely to cause side effects. However, whether the peak efficacy and toxicity occur at C
max is dependent on the pharmacodynamics profile of the drug. For example, toxicity and efficacy of drugs such as vigabatrin, which inhibits GABA transaminase, may have a poor correlation with serum concentration of the drug.
One of the most important properties of a drug to know is clearance. Clearance represents the body’s ability to eliminate drug completely from a particular volume of fluid (usually plasma) per unit of time. Clearance can be defined for particular organs, such as the liver or kidney. Total clearance represents the sum of the various organ clearances resulting in drug elimination
(e.g., hepatic and renal clearance). Total clearance determines steady-state serum blood concentration:
Concentration = Bioavailability × Dose/Total clearance
Clearance represents a good parameter by which to evaluate age-related changes in drug elimination. Although half-life, the period over which the concentration of the drug decreases to half of its original concentration in serum, is an important pharmacokinetic feature, it is dependent on the volume of distribution (VD), which is a measure of how the drug is distributed in the body (between blood and tissues, in particular). VD is the amount of drug in the body divided by the concentration in the blood. Drugs that are highly lipid soluble have a very high volume of distribution, whereas drugs that are lipid insoluble remain in the blood and have a low VD. Half-life is defined as
T1/2 = 0.693 VD
Total clearance Cls
Because VD varies significantly as a function of age and body distribution of fat, it is not a good measure of longitudinal changes in clearance of the drug from the body. However, half-life is a valuable practical pharmacokinetic measure.
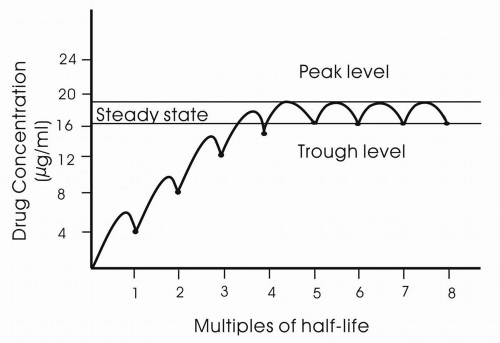
Steady state is the condition in which the drug is in a dynamic equilibrium in which the rate of clearance is equal to the rate of administration. Drugs will accumulate within the body if the drug has not been fully eliminated before the next dose. Steady-state concentration is reached after four to five half-lives (
Fig. 11-4).
Antiepileptic drugs can be excreted unchanged in the urine or be metabolized in the liver through the cytochrome (Cyp) P450 system (CYP1A2, CYP2A6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A) or via uridine 5′-diphosphate-glucuronosyltransferase. The majority of antiepileptic drugs that are metabolized in the liver go through the Cyp system. Lamotrigine is primarily metabolized via uridine 5′-diphosphate-glucuronosyltransferase.
Table 11-1 lists the effects of antiepileptic drugs on the Cyp system, and
Table 11-2 lists the antiepileptic drugs that are metabolized in the liver or excreted in the urine.
As a general rule, neonates and infants have slow hepatic metabolism, whereas hepatic metabolism is increased in children. Likewise, renal clearance of drugs is lower than that in adults, whereas renal clearance is greater in children than adults.
A constant fraction of the drug in the body is eliminated per unit time. The rate of elimination is proportional to the amount of drug in the body. The majority of drugs are eliminated in this way. When the rate of elimination is proportional to the amount of drug in the body, the drug has linear kinetics. For most antiepileptic drugs, the kinetics are linear. However, with phenytoin, when the enzymes responsible for its metabolism become saturated, the rate of elimination, in terms of the amount of the drug eliminated in a given period, does not increase in response to an increase in concentration. This is termed zero-order kinetics (
Fig. 11-5).
V. CARBAMAZEPINE (TEGRETOL)
B. Indications, Advantages, and Disadvantages
Carbamazepine is indicated as initial or adjunctive therapy for complex partial and tonic-clonic seizures. It was found to be one of the two safest and most effective drugs for these seizure types in comparative studies of older drugs (phenytoin is the other.) It is relatively nonsedating (sedation is mild and similar to that of phenytoin) and does not have the cosmetic side effects of phenytoin.
A loading dose of carbamazepine cannot be administered by any route. The drug must be given in divided doses. Carbamazepine must be started at a low dose, and the dosage must be built up over time. Carbamazepine may exacerbate absence seizures in some patients. An increased risk exists of spina bifida in infants born to mothers taking carbamazepine during pregnancy.
C. Pharmacokinetics
Approximately 75% to 85% of an orally administered dose of brand-name Tegretol, 200-mg tablets, is slowly absorbed after oral administration. The bioavailability of generic carbamazepine is sometimes less than that of brand-name Tegretol. Carbamazepine is 70% to 80% protein-bound. Carbamazepine is metabolized by the liver into 32 or more metabolites, some of which (especially carbamazepine epoxide) possess antiepileptic activity. Carbamazepine biotransformation exhibits timedependent pharmacokinetics (self-induction), which means that the plasma concentration may decrease unexpectedly during the first 2 months of administration when self-induction occurs.
D. Usual Pediatric and Adult Dosages
E. Formulations
Carbamazepine is marketed as standard 200-mg tablets (both brand-name Tegretol and generic), 100-mg chewable tablets (Tegretol), and suspension of 100 mg per 5 mL. These formulations are labeled for t.i.d. or q.i.d. administration.
Two sustained-release carbamazepine formulations (Carbatrol and Tegretol-XR) are available for b.i.d. administration. Although the two formulations use different technologies, they both perform well with b.i.d. administration. Sustained-release preparations may improve compliance, reduce toxicity associated with peak levels, and reduce breakthrough seizures at times of trough blood levels. Carbatrol is available as 200-mg and 300-mg capsules. Tegretol-XR is available as 100-mg, 200-mg, and 400-mg tablets. Sustained-release formulations of carbamazepine are the preferred mode of administration.
F. Toxicity
Local toxicity consists of gastric irritability that is usually managed if the patient takes the drug after meals. Common dose-related toxicity includes diplopia or blurred vision, dizziness, drowsiness, ataxia, and headache. At high plasma concentrations, the following may occur: tremor, dystonia, chorea, depression, irritability, psychosis, convulsions, water retention (inappropriate antidiuretic hormone-like syndrome), congestive heart failure, and cardiac arrhythmias.
Idiosyncratic toxicities are rash (common) and, more rarely, anemia, agranulocytosis, leukopenia, thrombocytopenia, hypersensitivity syndrome (dermatitis, eosinophilia, lymphadenopathy, splenomegaly), and cholestatic and hepatocellular jaundice. The rate of fatal idiosyncratic reactions with carbamazepine is
estimated currently at 1 in 100,000 to 200,000 patients. Although this factor is a matter of concern, the risk is in a range similar to that of other commonly used drugs, such as penicillin.
The issue of reduced bone marrow density is discussed in
Chapter 10.
G. Drug Interactions
Adding carbamazepine may increase the plasma concentration of fluoxetine, phenytoin, and tricyclic antidepressants. Adding carbamazepine may reduce the plasma concentrations of antiviral and anticancer drugs metabolized by CYP450, benzodiazepines, corticosteroids, cyclosporine, felbamate, griseofulvin, haloperidol, lamotrigine, oral contraceptives, oxcarbazepine, theophylline, tiagabine, topiramate, valproic acid, warfarin, and zonisamide. Propoxyphene, erythromycin, clarithromycin, chloramphenicol, isoniazid, verapamil, grapefruit juice, and cimetidine may increase the carbamazepine plasma concentration, whereas phenobarbital, phenytoin, felbamate, Saint John’s wort, and primidone may reduce the carbamazepine plasma concentration. Adding valproic acid to carbamazepine may increase plasma carbamazepine epoxide concentration. Adding lamotrigine to carbamazepine may enhance the usual neurotoxic side effects of carbamazepine.
H. Disease States
Carbamazepine may precipitate or enhance congestive heart failure. Its use should be avoided in this setting or in cases in which major arrhythmias are a concern. Plasma concentrations and potential toxicity must be closely watched when the drug is used in patients with renal or hepatic disease.
I. Pregnancy
A 0.5% to 1% risk of spina bifida exists in infants born to mothers who took carbamazepine during the first trimester. Reports are found of increased risk of other congenital malformations in infants born to mothers taking antiepileptic drugs. The potential risks and benefits of using this drug in pregnancy must be carefully weighed for each patient. Because alternative antiepileptic drugs do not increase the risk of spina bifida, the authors generally prefer to use one of the alternative drugs (any other antiepileptic drug except valproic acid) in women of childbearing potential.
The drug is excreted in breast milk. The risks of exposure of the infant to the drug are unknown. The benefits of breast-feeding, the risk to the infant of drug exposure, and the risk to the mother of stopping the drug must be weighed in each case.