PART 10: Disorders of the Cardiovascular System
SECTION 1 | INTRODUCTION TO CARDIOVASCULAR DISORDERS |
264 | Approach to the Patient with Possible Cardiovascular Disease |
THE MAGNITUDE OF THE PROBLEM
Cardiovascular diseases comprise the most prevalent serious disorders in industrialized nations and are a rapidly growing problem in developing nations (Chap. 266e). Age-adjusted death rates for coronary heart disease have declined by two-thirds in the last four decades in the United States, reflecting the identification and reduction of risk factors as well as improved treatments and interventions for the management of coronary artery disease, arrhythmias, and heart failure. Nonetheless, cardiovascular diseases remain the most common causes of death, responsible for 35% of all deaths, almost 1 million deaths each year. Approximately one-fourth of these deaths are sudden. In addition, cardiovascular diseases are highly prevalent, diagnosed in 80 million adults, or ~35% of the adult population. The growing prevalence of obesity (Chap. 416), type 2 diabetes mellitus (Chap. 417), and metabolic syndrome (Chap. 422), which are important risk factors for atherosclerosis, now threatens to reverse the progress that has been made in the age-adjusted reduction in the mortality rate of coronary heart disease.
For many years cardiovascular disease was considered to be more common in men than in women. In fact, the percentage of all deaths secondary to cardiovascular disease is higher among women (43%) than among men (37%) (Chap. 6e). In addition, although the absolute number of deaths secondary to cardiovascular disease has declined over the past decades in men, this number has actually risen in women. Inflammation, obesity, type 2 diabetes mellitus, and the metabolic syndrome appear to play more prominent roles in the development of coronary atherosclerosis in women than in men. Coronary artery disease (CAD) is more frequently associated with dysfunction of the coronary microcirculation in women than in men. Exercise electrocardiography has a lower diagnostic accuracy in the prediction of epicardial obstruction in women than in men.
NATURAL HISTORY
Cardiovascular disorders often present acutely, as in a previously asymptomatic person who develops an acute myocardial infarction (Chap. 295), or a previously asymptomatic patient with hypertrophic cardiomyopathy (Chap. 287), or with a prolonged QT interval (Chap. 277) whose first clinical manifestation is syncope or even sudden death. However, the alert physician may recognize the patient at risk for these complications long before they occur and often can take measures to prevent their occurrence. For example, a patient with acute myocardial infarction will often have had risk factors for atherosclerosis for many years. Had these risk factors been recognized, their elimination or reduction might have delayed or even prevented the infarction. Similarly, a patient with hypertrophic cardiomyopathy may have had a heart murmur for years and a family history of this disorder. These findings could have led to an echocardiographic examination, recognition of the condition, and appropriate therapy long before the occurrence of a serious acute manifestation.
Patients with valvular heart disease or idiopathic dilated cardiomyopathy, by contrast, may have a prolonged course of gradually increasing dyspnea and other manifestations of chronic heart failure that is punctuated by episodes of acute deterioration only late in the course of the disease. Understanding the natural history of various cardiac disorders is essential for applying appropriate diagnostic and therapeutic measures to each stage of the condition, as well as for providing the patient and family with the likely prognosis.
CARDIAC SYMPTOMS
The symptoms caused by heart disease result most commonly from myocardial ischemia, disturbance of the contraction and/or relaxation of the myocardium, obstruction to blood flow, or an abnormal cardiac rhythm or rate. Ischemia, which is caused by an imbalance between the heart’s oxygen supply and demand, is manifest most frequently as chest discomfort (Chap. 19), whereas reduction of the pumping ability of the heart commonly leads to fatigue and elevated intravascular pressure upstream of the failing ventricle. The latter results in abnormal fluid accumulation, with peripheral edema (Chap. 50) or pulmonary congestion and dyspnea (Chap. 47e). Obstruction to blood flow, as occurs in valvular stenosis, can cause symptoms resembling those of myocardial failure (Chap. 279). Cardiac arrhythmias often develop suddenly, and the resulting symptoms and signs—palpitations (Chap. 52), dyspnea, hypotension, and syncope (Chap. 27)—generally occur abruptly and may disappear as rapidly as they develop.
Although dyspnea, chest discomfort, edema, and syncope are cardinal manifestations of cardiac disease, they occur in other conditions as well. Thus, dyspnea is observed in disorders as diverse as pulmonary disease, marked obesity, and anxiety (Chap. 47e). Similarly, chest discomfort may result from a variety of noncardiac and cardiac causes other than myocardial ischemia (Chap. 19). Edema, an important finding in untreated or inadequately treated heart failure, also may occur with primary renal disease and in hepatic cirrhosis (Chap. 50). Syncope occurs not only with serious cardiac arrhythmias but in a number of neurologic conditions as well (Chap. 27). Whether heart disease is responsible for these symptoms frequently can be determined by carrying out a careful clinical examination (Chap. 267), supplemented by noninvasive testing using electrocardiography at rest and during exercise (Chap. 268), echocardiography, roentgenography, and other forms of myocardial imaging (Chap. 270e).
Myocardial or coronary function that may be adequate at rest may be insufficient during exertion. Thus, dyspnea and/or chest discomfort that appear during activity are characteristic of patients with heart disease, whereas the opposite pattern, i.e., the appearance of these symptoms at rest and their remission during exertion, is rarely observed in such patients. It is important, therefore, to question the patient carefully about the relation of symptoms to exertion.
Many patients with cardiovascular disease may be asymptomatic both at rest and during exertion but may present with an abnormal physical finding such as a heart murmur, elevated arterial pressure, or an abnormality of the electrocardiogram (ECG) or imaging test. It is important to assess the global risk of CAD in asymptomatic individuals, using a combination of clinical assessment and measurement of cholesterol and its fractions, as well as other biomarkers, such as C-reactive protein, in some patients (Chap. 291e). Since the first clinical manifestation of CAD may be catastrophic—sudden cardiac death, acute myocardial infarction, or stroke in previous asymptomatic persons—it is mandatory to identify those at high risk of such events and institute further testing and preventive measures.
DIAGNOSIS
As outlined by the New York Heart Association (NYHA), the elements of a complete cardiac diagnosis include the systematic consideration of the following:
1. The underlying etiology. Is the disease congenital, hypertensive, ischemic, or inflammatory in origin?
2. The anatomic abnormalities. Which chambers are involved? Are they hypertrophied, dilated, or both? Which valves are affected? Are they regurgitant and/or stenotic? Is there pericardial involvement? Has there been a myocardial infarction?
3. The physiologic disturbances. Is an arrhythmia present? Is there evidence of congestive heart failure or myocardial ischemia?
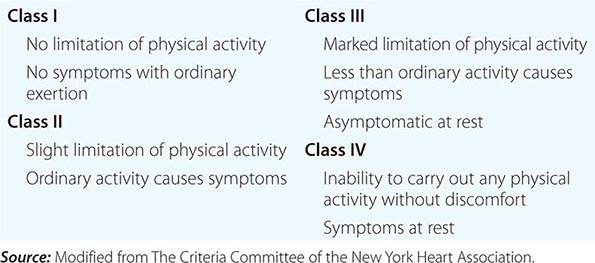
4. Functional disability. How strenuous is the physical activity required to elicit symptoms? The classification provided by the NYHA has been found to be useful in describing functional disability (Table 264-1).
NEW YORK HEART ASSOCIATION FUNCTIONAL CLASSIFICATION |
One example may serve to illustrate the importance of establishing a complete diagnosis. In a patient who presents with exertional chest discomfort, the identification of myocardial ischemia as the etiology is of great clinical importance. However, the simple recognition of ischemia is insufficient to formulate a therapeutic strategy or prognosis until the underlying anatomic abnormalities responsible for the myocardial ischemia, e.g., coronary atherosclerosis or aortic stenosis, are identified and a judgment is made about whether other physiologic disturbances that cause an imbalance between myocardial oxygen supply and demand, such as severe anemia, thyrotoxicosis, or supraventricular tachycardia, play contributory roles. Finally, the severity of the disability should govern the extent and tempo of the workup and strongly influence the therapeutic strategy that is selected.
The establishment of a correct and complete cardiac diagnosis usually commences with the history and physical examination (Chap. 267). Indeed, the clinical examination remains the basis for the diagnosis of a wide variety of disorders. The clinical examination may then be supplemented by five types of laboratory tests: (1) ECG (Chap. 268), (2) noninvasive imaging examinations (chest roentgenogram, echocardiogram, radionuclide imaging, computed tomographic imaging, positron emission tomography, and magnetic resonance imaging) (Chap. 270e), (3) blood tests to assess risk (e.g., lipid determinations, C-reactive protein [Chap. 291e]) or cardiac function (e.g., brain natriuretic peptide [BNP] [Chap. 279]), (4) occasionally specialized invasive examinations (i.e., cardiac catheterization and coronary arteriography [Chap. 272]), and (5) genetic tests to identify monogenic cardiac diseases (e.g., hypertrophic cardiomyopathy [Chap. 287], Marfan’s syndrome [Chap. 427], and abnormalities of cardiac ion channels that lead to prolongation of the QT interval and an increase in the risk of sudden death [Chap. 276]). These tests are becoming more widely available.
FAMILY HISTORY
In eliciting the history of a patient with known or suspected cardiovascular disease, particular attention should be directed to the family history. Familial clustering is common in many forms of heart disease. Mendelian transmission of single-gene defects may occur, as in hypertrophic cardiomyopathy (Chap. 287), Marfan’s syndrome (Chap. 427), and sudden death associated with a prolonged QT syndrome (Chap. 277). Premature coronary disease and essential hypertension, type 2 diabetes mellitus, and hyperlipidemia (the most important risk factors for CAD) are usually polygenic disorders. Although familial transmission may be less obvious than in the monogenic disorders, it is helpful in assessing risk and prognosis in polygenic disorders, as well. Familial clustering of cardiovascular diseases not only may occur on a genetic basis but also may be related to familial dietary or behavior patterns, such as excessive ingestion of salt or calories and cigarette smoking.
ASSESSMENT OF FUNCTIONAL IMPAIRMENT
When an attempt is made to determine the severity of functional impairment in a patient with heart disease, it is helpful to ascertain the level of activity and the rate at which it is performed before symptoms develop. Thus, it is not sufficient to state that the patient complains of dyspnea. The breathlessness that occurs after running up two long flights of stairs denotes far less functional impairment than do similar symptoms that occur after taking a few steps on level ground. Also, the degree of customary physical activity at work and during recreation should be considered. The development of two-flight dyspnea in a well-conditioned marathon runner may be far more significant than the development of one-flight dyspnea in a previously sedentary person. The history should include a detailed consideration of the patient’s therapeutic regimen. For example, the persistence or development of edema, breathlessness, and other manifestations of heart failure in a patient who is receiving optimal doses of diuretics and other therapies for heart failure (Chap. 279) is far graver than are similar manifestations in the absence of treatment. Similarly, the presence of angina pectoris despite treatment with optimal doses of multiple antianginal drugs (Chap. 293) is more serious than it is in a patient on no therapy. In an effort to determine the progression of symptoms, and thus the severity of the underlying illness, it may be useful to ascertain what, if any, specific tasks the patient could have carried out 6 months or 1 year earlier that he or she cannot carry out at present.
ELECTROCARDIOGRAM
(See also Chap. 268) Although an ECG usually should be recorded in patients with known or suspected heart disease, with the exception of the identification of arrhythmias, conduction abnormalities, ventricular hypertrophy, and acute myocardial infarction, it generally does not establish a specific diagnosis. The range of normal electrocardiographic findings is wide, and the tracing can be affected significantly by many noncardiac factors, such as age, body habitus, and serum electrolyte concentrations. In general, electrocardiographic changes should be interpreted in the context of other abnormal cardiovascular findings.
ASSESSMENT OF THE PATIENT WITH A HEART MURMUR
(Fig. 264-1) The cause of a heart murmur can often be readily elucidated from a systematic evaluation of its major attributes: timing, duration, intensity, quality, frequency, configuration, location, and radiation when considered in the light of the history, general physical examination, and other features of the cardiac examination, as described in Chap. 267.
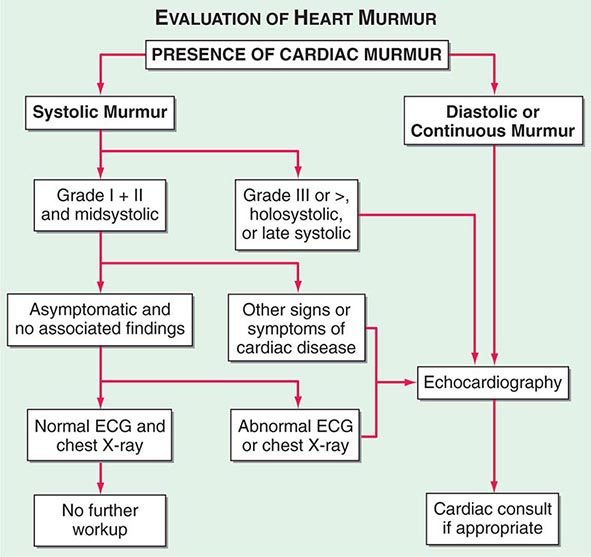
FIGURE 264-1 Approach to the evaluation of a heart murmur. ECG, electrocardiogram. (From RA O’Rourke, in Primary Cardiology, 2nd ed, E Braunwald, L Goldman [eds]. Philadelphia, Saunders, 2003.)
The majority of heart murmurs are midsystolic and soft (grades I–II/VI). When such a murmur occurs in an asymptomatic child or young adult without other evidence of heart disease on clinical examination, it is usually benign and echocardiography generally is not required. By contrast, two-dimensional and Doppler echocardiography (Chap. 270e) are indicated in patients with loud systolic murmurs (grades ≥III/VI), especially those that are holosystolic or late systolic, and in most patients with diastolic or continuous murmurs.
PITFALLS IN CARDIOVASCULAR MEDICINE
Increasing subspecialization in internal medicine and the perfection of advanced diagnostic techniques in cardiology can lead to several undesirable consequences. Examples include the following:
1. Failure by the noncardiologist to recognize important cardiac manifestations of systemic illnesses. For example, the presence of mitral stenosis, patent foramen ovale, and/or transient atrial arrhythmia should be considered in a patient with stroke, or the presence of pulmonary hypertension and cor pulmonale should be considered in a patient with scleroderma or Raynaud’s syndrome. A cardiovascular examination should be carried out to identify and estimate the severity of the cardiovascular involvement that accompanies many noncardiac disorders.
2. Failure by the cardiologist to recognize underlying systemic disorders in patients with heart disease. For example, hyperthyroidism should be considered in an elderly patient with atrial fibrillation and unexplained heart failure, and Lyme disease should be considered in a patient with unexplained fluctuating atrioventricular block. A cardiovascular abnormality may provide the clue critical to the recognition of some systemic disorders. For example, an unexplained pericardial effusion may provide an early clue to the diagnosis of tuberculosis or a neoplasm.
3. Overreliance on and overutilization of laboratory tests, particularly invasive techniques, for the evaluation of the cardiovascular system. Cardiac catheterization and coronary arteriography (Chap. 272) provide precise diagnostic information that may be crucial in developing a therapeutic plan in patients with known or suspected CAD. Although a great deal of attention has been directed to these examinations, it is important to recognize that they serve to supplement, not supplant, a careful examination carried out with clinical and noninvasive techniques. A coronary arteriogram should not be performed in lieu of a careful history in patients with chest pain suspected of having ischemic heart disease. Although coronary arteriography may establish whether the coronary arteries are obstructed and to what extent, the results of the procedure by themselves often do not provide a definitive answer to the question of whether a patient’s complaint of chest discomfort is attributable to coronary atherosclerosis and whether or not revascularization is indicated.
Despite the value of invasive tests in certain circumstances, they entail some small risk to the patient, involve discomfort and substantial cost, and place a strain on medical facilities. Therefore, they should be carried out only if the results can be expected to modify the patient’s management.
DISEASE PREVENTION AND MANAGEMENT
The prevention of heart disease, especially of CAD, is one of the most important tasks of primary care health givers as well as cardiologists. Prevention begins with risk assessment, followed by attention to lifestyle, such as achieving optimal weight, physical activity, and smoking cessation, and then aggressive treatment of all abnormal risk factors, such as hypertension, hyperlipidemia, and diabetes mellitus (Chap. 417).
After a complete diagnosis has been established in patients with known heart disease, a number of management options are usually available. Several examples may be used to demonstrate some of the principles of cardiovascular therapeutics:
1. In the absence of evidence of heart disease, the patient should be clearly informed of this assessment and not be asked to return at intervals for repeated examinations. If there is no evidence of disease, such continued attention may lead to the patient’s developing inappropriate concern about the possibility of heart disease.
2. If there is no evidence of cardiovascular disease but the patient has one or more risk factors for the development of ischemic heart disease (Chap. 293), a plan for their reduction should be developed and the patient should be retested at intervals to assess compliance and efficacy in risk reduction.
3. Asymptomatic or mildly symptomatic patients with valvular heart disease that is anatomically severe should be evaluated periodically, every 6 to 12 months, by clinical and noninvasive examinations. Early signs of deterioration of ventricular function may signify the need for surgical treatment before the development of disabling symptoms, irreversible myocardial damage, and excessive risk of surgical treatment (Chap. 283).
4. In patients with CAD (Chap. 293), available practice guidelines should be considered in the decision on the form of treatment (medical, percutaneous coronary intervention, or surgical revascularization). Mechanical revascularization may be employed too frequently in the United States and too infrequently in Eastern Europe and developing nations. The mere presence of angina pectoris and/or the demonstration of critical coronary arterial narrowing at angiography should not reflexively evoke a decision to treat the patient by revascularization. Instead, these interventions should be limited to patients with CAD whose angina has not responded adequately to medical treatment or in whom revascularization has been shown to improve the natural history (e.g., acute coronary syndrome or multivessel CAD with left ventricular dysfunction).
265e | Basic Biology of the Cardiovascular System |
THE BLOOD VESSEL
VASCULAR ULTRASTRUCTURE
Blood vessels participate in homeostasis on a moment-to-moment basis and contribute to the pathophysiology of diseases of virtually every organ system. Hence, an understanding of the fundamentals of vascular biology furnishes a foundation for understanding the normal function of all organ systems and many diseases. The smallest blood vessels—capillaries—consist of a monolayer of endothelial cells apposed to a basement membrane, adjacent to occasional smooth-muscle-like cells known as pericytes (Fig. 265e-1A). Unlike larger vessels, pericytes do not invest the entire microvessel to form a continuous sheath. Arteries typically have a trilaminar structure (Fig. 265e-1B–E). The intima consists of a monolayer of endothelial cells continuous with those of the capillaries. The middle layer, or tunica media, consists of layers of smooth-muscle cells; in veins, the media can contain just a few layers of smooth-muscle cells (Fig. 265e-1B). The outer layer, the adventitia, consists of looser extracellular matrix with occasional fibroblasts, mast cells, and nerve terminals. Larger arteries have their own vasculature, the vasa vasorum, which nourishes the outer aspects of the tunica media. The adventitia of many veins surpasses the intima in thickness.
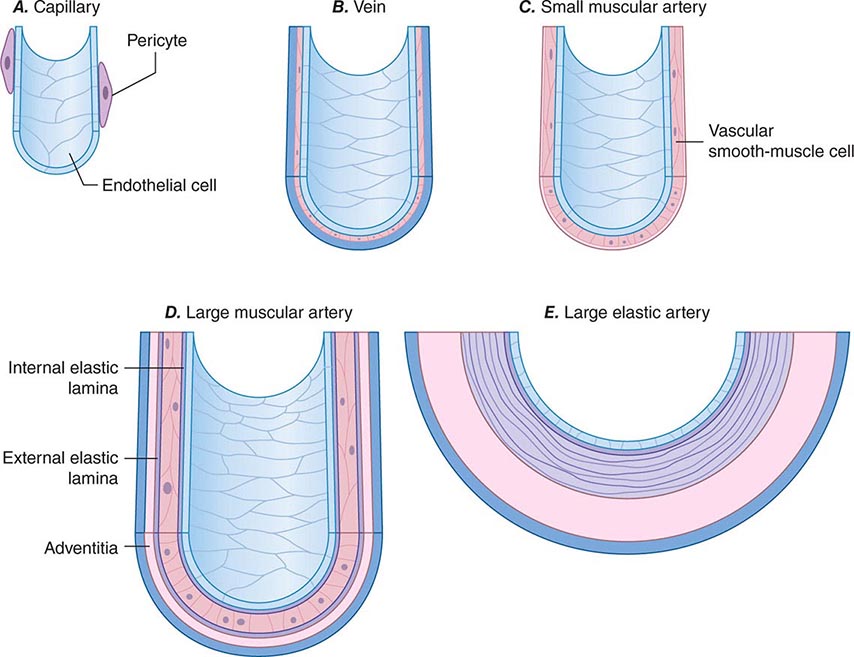
FIGURE 265e-1 Schematics of the structures of various types of blood vessels. A. Capillaries consist of an endothelial tube in contact with a discontinuous population of pericytes. B. Veins typically have thin medias and thicker adventitias. C. A small muscular artery features a prominent tunica media. D. Larger muscular arteries have a prominent media with smooth-muscle cells embedded in a complex extracellular matrix. E. Larger elastic arteries have cylindrical layers of elastic tissue alternating with concentric rings of smooth-muscle cells.
The tone of muscular arterioles regulates blood pressure and flow through various arterial beds. These smaller arteries have a relatively thick tunica media in relation to the adventitia (Fig. 265e-1C). Medium-size muscular arteries similarly contain a prominent tunica media (Fig. 265e-1D); atherosclerosis commonly affects this type of muscular artery. The larger elastic arteries have a much more structured tunica media consisting of concentric bands of smooth-muscle cells, interspersed with strata of elastin-rich extracellular matrix sandwiched between layers of smooth-muscle cells (Fig. 265e-1E). Larger arteries have a clearly demarcated internal elastic lamina that forms the barrier between the intima and the media. An external elastic lamina demarcates the media of arteries from the surrounding adventitia.
ORIGIN OF VASCULAR CELLS
The intima in human arteries often contains occasional resident smooth-muscle cells beneath the monolayer of vascular endothelial cells. The embryonic origin of smooth-muscle cells in various types of artery differs. Some upper-body arterial smooth-muscle cells derive from the neural crest, whereas lower-body arteries generally recruit smooth-muscle cells from neighboring mesodermal structures during development. Derivatives of the proepicardial organ, which gives rise to the epicardial layer of the heart, contribute to the vascular smooth-muscle cells of the coronary arteries. Bone marrow–derived endothelial progenitors may aid repair of damaged or aging arteries. In addition, multipotent vascular stem cells resident in vessel walls may give rise to the smooth-muscle cells that accumulate in injured or atheromatous arteries (Chaps. 88, 89e, and 90e).
VASCULAR CELL BIOLOGY
Endothelial Cell The key cell of the vascular intima, the endothelial cell, has manifold functions in health and disease. The endothelium forms the interface between tissues and the blood compartment. It therefore must regulate the entry of molecules and cells into tissues in a selective manner. The ability of endothelial cells to serve as a selectively permeable barrier fails in many vascular disorders, including atherosclerosis, hypertension, and renal disease. This dysregulation of permeability also occurs in pulmonary edema and other situations of “capillary leak.”
The endothelium also participates in the local regulation of blood flow and vascular caliber. Endogenous substances produced by endothelial cells such as prostacyclin, endothelium-derived hyperpolarizing factor, nitric oxide (NO), and hydrogen peroxide (H2O2) provide tonic vasodilatory stimuli under physiologic conditions in vivo (Table 265e-1). Impaired production or excess catabolism of NO impairs this endothelium-dependent vasodilator function and may contribute to excessive vasoconstriction in various pathologic situations. Measurement of flow-mediated dilatation can assess endothelial vasodilator function in humans (Fig. 265e-2). By contrast, endothelial cells also produce potent vasoconstrictor substances such as endothelin in a regulated fashion. Excessive production of reactive oxygen species, such as superoxide anion (O2–), by endothelial or smooth-muscle cells under pathologic conditions (e.g., excessive exposure to angiotensin II), can promote local oxidative stress and inactivate NO.
ENDOTHELIAL FUNCTIONS IN HEALTH AND DISEASE |
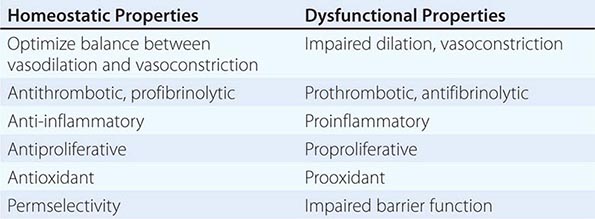
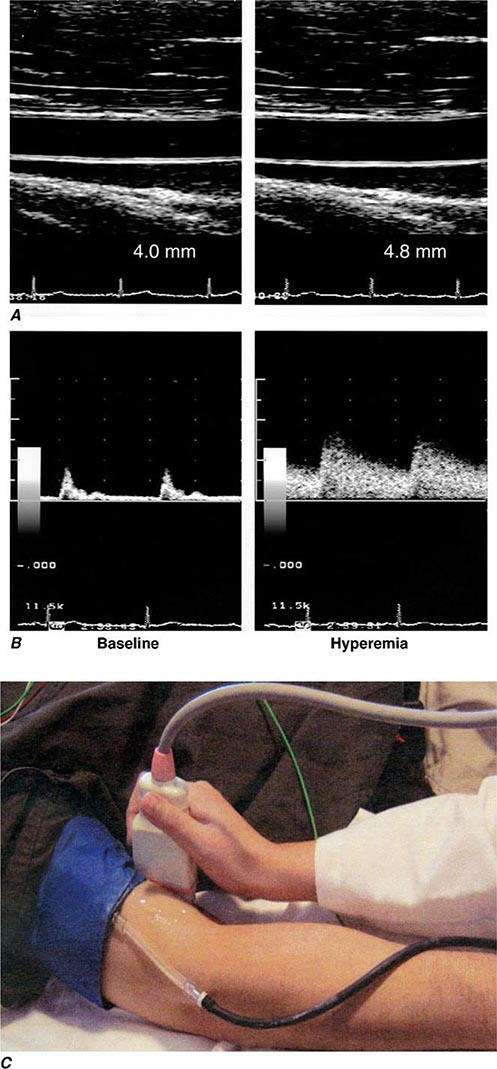
FIGURE 265e-2 Assessment of endothelial function in vivo using blood pressure cuff occlusion and release. Upon deflation of the cuff, an ultrasound probe monitors changes in diameter (A) and blood flow (B) of the brachial artery (C). (Reproduced with permission of J. Vita, MD.)
The endothelial monolayer contributes critically to inflammatory processes involved in normal host defenses and pathologic states. The normal endothelium resists prolonged contact with blood leukocytes; however, when activated by bacterial products such as endotoxin or by proinflammatory cytokines released during infection or injury, endothelial cells express an array of leukocyte adhesion molecules that bind various classes of leukocytes. The endothelial cells appear to recruit selectively different classes of leukocytes in different pathologic conditions. The gamut of adhesion molecules and chemokines generated during acute bacterial infection tends to recruit granulocytes. In chronic inflammatory diseases such as tuberculosis and atherosclerosis, endothelial cells express adhesion molecules that favor the recruitment of mononuclear leukocytes that characteristically accumulate in these conditions.
The endothelium also dynamically regulates thrombosis and hemostasis. NO, in addition to its vasodilatory properties, can limit platelet activation and aggregation. Like NO, prostacyclin produced by endothelial cells under normal conditions not only provides a vasodilatory stimulus but also antagonizes platelet activation and aggregation. Thrombomodulin expressed on the surface of endothelial cells binds thrombin at low concentrations and inhibits coagulation through activation of the protein C pathway, inactivating clotting factors Va and VIIIa and thus combating thrombus formation. The surface of endothelial cells contains heparan sulfate glycosaminoglycans that furnish an endogenous antithrombotic coating to the vasculature. Endothelial cells also participate actively in fibrinolysis and its regulation. They express receptors for plasminogen and plasminogen activators and produce tissue-type plasminogen activator. Through local generation of plasmin, the normal endothelial monolayer can promote the lysis of nascent thrombi.
When activated by inflammatory cytokines, bacterial endotoxin, or angiotensin II, for example, endothelial cells can produce substantial quantities of the major inhibitor of fibrinolysis, plasminogen activator inhibitor 1 (PAI-1). Thus, in pathologic circumstances, the endothelial cell may promote local thrombus accumulation rather than combat it. Inflammatory stimuli also induce the expression of the potent procoagulant tissue factor, a contributor to disseminated intravascular coagulation in sepsis.
Endothelial cells also participate in the pathophysiology of a number of immune-mediated diseases. Lysis of endothelial cells mediated by complement provides an example of immunologically mediated tissue injury. The presentation of foreign histocompatibility complex antigens by endothelial cells in solid-organ allografts can promote allograft arteriopathy. In addition, immune-mediated endothelial injury may contribute in some patients with thrombotic thrombocytopenic purpura and patients with hemolytic-uremic syndrome. Thus, in addition to the involvement of innate immune responses, endothelial cells participate actively in both humoral and cellular limbs of the immune response.
Endothelial cells regulate growth of subjacent smooth-muscle cells. Heparan sulfate glycosaminoglycans elaborated by endothelial cells can inhibit smooth-muscle proliferation. In contrast, when exposed to various injurious stimuli, endothelial cells can elaborate growth factors and chemoattractants, such as platelet-derived growth factor, that can promote the migration and proliferation of vascular smooth-muscle cells. Dysregulated elaboration of these growth-stimulatory molecules may promote smooth-muscle accumulation in atherosclerotic lesions.
Vascular Smooth-Muscle Cell The vascular smooth-muscle cell, the major cell type of the media layer of blood vessels, also contributes actively to vascular pathobiology. Contraction and relaxation of smooth-muscle cells at the level of the muscular arteries controls blood pressure and, hence, regional blood flow and the afterload experienced by the left ventricle (see below). The vasomotor tone of veins, which is governed by smooth-muscle cell tone, regulates the capacitance of the venous tree and influences the preload experienced by both ventricles. Smooth-muscle cells in the adult vessel seldom replicate in the absence of arterial injury or inflammatory activation. Proliferation and migration of arterial smooth-muscle cells, associated with functional modulation characterized by lower content of contractile proteins and greater production of extracellular matrix macromolecules, can contribute to the development of arterial stenoses in atherosclerosis, arteriolar remodeling that can sustain and propagate hypertension, and the hyperplastic response of arteries injured by percutaneous intervention. In the pulmonary circulation, smooth-muscle migration and proliferation contribute decisively to the pulmonary vascular disease that gradually occurs in response to sustained high-flow states such as left-to-right shunts. Such pulmonary vascular disease provides a major obstacle to the management of many patients with adult congenital heart disease. Among other mediators, microRNAs have emerged as powerful regulators of this transition, offering new targets for intervention.
Smooth-muscle cells secrete the bulk of vascular extracellular matrix. Excessive production of collagen and glycosaminoglycans contributes to the remodeling and altered functions and biomechanics of arteries affected by hypertension or atherosclerosis. In larger elastic arteries, the elastin synthesized by smooth-muscle cells serves to maintain not only normal arterial structure, but also hemodynamic function. The ability of the larger arteries, such as the aorta, to store the kinetic energy of systole promotes tissue perfusion during diastole. Arterial stiffness associated with aging or disease, as manifested by a widening pulse pressure, increases left ventricular afterload and portends a poor outcome.
Like endothelial cells, vascular smooth-muscle cells do not merely respond to vasomotor or inflammatory stimuli elaborated by other cell types but can themselves serve as a source of such stimuli. For example, when exposed to bacterial endotoxin or other proinflammatory stimuli, smooth-muscle cells can elaborate cytokines and other inflammatory mediators. Like endothelial cells, upon inflammatory activation, arterial smooth-muscle cells can produce prothrombotic mediators such as tissue factor, the antifibrinolytic protein PAI-1, and other molecules that modulate thrombosis and fibrinolysis. Smooth-muscle cells also elaborate autocrine growth factors that can amplify hyperplastic responses to arterial injury.
Vascular Smooth-Muscle Cell Function Vascular smooth-muscle cells govern vessel tone. Those cells contract when stimulated by a rise in intracellular calcium concentration by calcium influx through the plasma membrane and by calcium release from intracellular stores (Fig. 265e-3). In vascular smooth-muscle cells, voltage-dependent L-type calcium channels open with membrane depolarization, which is regulated by energy-dependent ion pumps such as the Na+,K+-ATPase pump and ion channels such as the Ca2+-sensitive K+ channel. Local changes in intracellular calcium concentration, termed calcium sparks, result from the influx of calcium through the voltage-dependent calcium channel and are caused by the coordinated activation of a cluster of ryanodine-sensitive calcium release channels in the sarcoplasmic reticulum (see below). Calcium sparks directly augment intracellular calcium concentration and indirectly increase intracellular calcium concentration by activating chloride channels. In addition, calcium sparks reduce smooth-muscle contractility by activating large-conductance calcium-sensitive K+ channels, hyperpolarizing the cell membrane and thereby limiting further voltage-dependent increases in intracellular calcium.
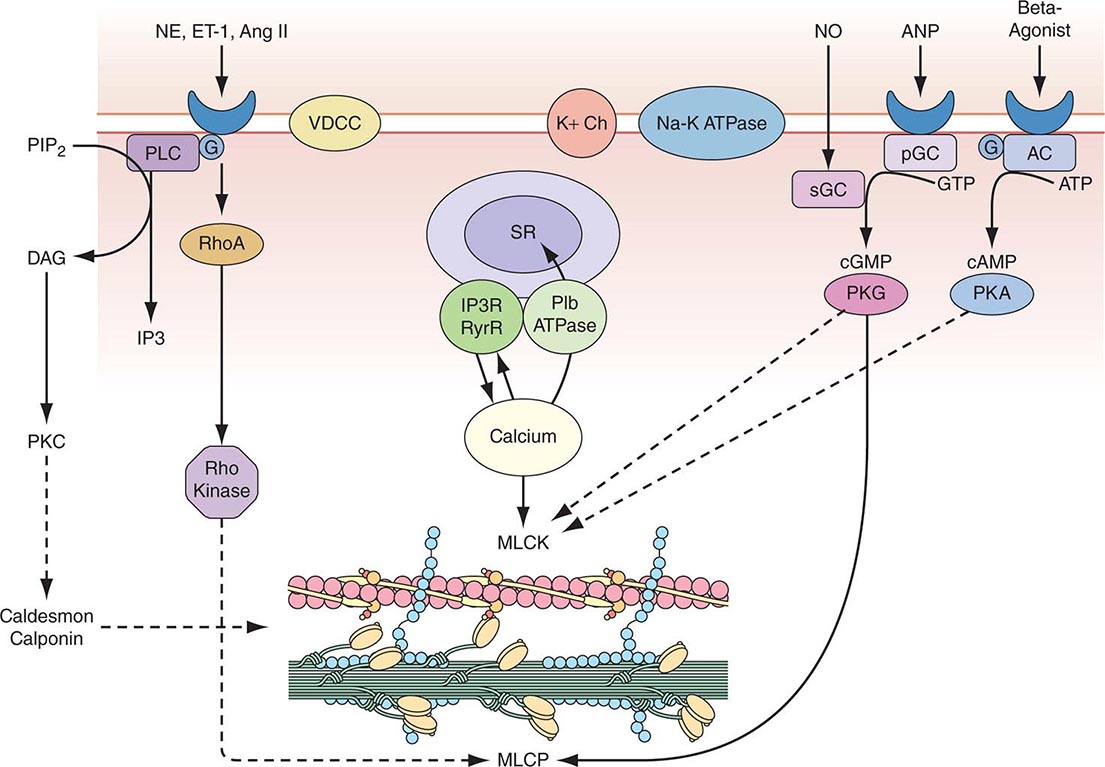
FIGURE 265e-3 Regulation of vascular smooth-muscle cell calcium concentration and actomyosin ATPase-dependent contraction. AC, adenylyl cyclase; Ang II, angiotensin II; ANP, atrial natriuretic peptide; DAG, diacylglycerol; ET-1, endothelin-1; G, G protein; IP3, inositol 1,4,5-trisphosphate; MLCK, myosin light chain kinase; MLCP, myosin light chain phosphatase; NE, norepinephrine; NO, nitric oxide; pGC, particular guanylyl cyclase; PIP2, phosphatidylinositol 4,5-bisphosphate; PKA, protein kinase A; PKC, protein kinase C; PKG, protein kinase G; PLC, phospholipase C; sGC, soluble guanylyl cyclase; SR, sarcoplasmic reticulum; VDCC, voltage-dependent calcium channel. (Modified from B Berk, in Vascular Medicine, 3rd ed. Philadelphia, Saunders, Elsevier, 2006, p. 23; with permission.)
Biochemical agonists also increase intracellular calcium concentration, in this case by receptor-dependent activation of phospholipase C with hydrolysis of phosphatidylinositol 4,5-bisphosphate, resulting in the generation of diacylglycerol (DAG) and inositol 1,4,5-trisphosphate (IP3). These membrane lipid derivatives in turn activate protein kinase C and increase intracellular calcium concentration. In addition, IP3 binds to specific receptors on the sarcoplasmic reticulum membrane to increase calcium efflux from this calcium storage pool into the cytoplasm.
Vascular smooth-muscle cell contraction depends principally on the phosphorylation of myosin light chain, which in the steady state, reflects the balance between the actions of myosin light chain kinase and myosin light chain phosphatase. Calcium activates myosin light chain kinase through the formation of a calcium-calmodulin complex. Phosphorylation of myosin light chain by this kinase augments myosin ATPase activity and enhances contraction. Myosin light chain phosphatase dephosphorylates myosin light chain, reducing myosin ATPase activity and contractile force. Phosphorylation of the myosin-binding subunit (thr695) of myosin light chain phosphatase by Rho kinase inhibits phosphatase activity and induces calcium sensitization of the contractile apparatus. Rho kinase is itself activated by the small GTPase RhoA, which is stimulated by guanosine exchange factors and inhibited by GTPase-activating proteins.
Both cyclic AMP and cyclic GMP relax vascular smooth-muscle cells through complex mechanisms. β agonists, acting through their G-protein-coupled receptors activate adenylyl cyclase to convert ATP to cyclic AMP; NO and atrial natriuretic peptide acting directly and via a G-protein-coupled receptor, respectively, activate guanylyl cyclase to convert GTP to cyclic GMP. These agents in turn activate protein kinase A and protein kinase G, respectively, which inactivate myosin light chain kinase and decrease vascular smooth-muscle cell tone. In addition, protein kinase G can interact directly with the myosin-binding substrate subunit of myosin light chain phosphatase, increasing phosphatase activity and decreasing vascular tone. Finally, several mechanisms drive NO-dependent, protein kinase G–mediated reductions in vascular smooth-muscle cell calcium concentration, including phosphorylation-dependent inactivation of RhoA; decreased IP3 formation; phosphorylation of the IP3 receptor–associated cyclic GMP kinase substrate, with subsequent inhibition of IP3 receptor function; phosphorylation of phospholamban, which increases calcium ATPase activity and sequestration of calcium in the sarcoplasmic reticulum; and protein kinase G–dependent stimulation of plasma membrane calcium ATPase activity, perhaps by activation of the Na+,K+-ATPase pump or hyperpolarization of the cell membrane by activation of calcium-dependent K+ channels.
Control of Vascular Smooth-Muscle Cell Tone The autonomic nervous system and endothelial cells modulate vascular smooth-muscle cells in a tightly regulated manner. Autonomic neurons enter the blood vessel medial layer from the adventitia and modulate vascular smooth-muscle cell tone in response to baroreceptors and chemoreceptors within the aortic arch and carotid bodies and in response to thermoreceptors in the skin. These regulatory components include rapidly acting reflex arcs modulated by central inputs that respond to sensory inputs (olfactory, visual, auditory, and tactile) as well as emotional stimuli. Three classes of nerves mediate autonomic regulation of vascular tone: sympathetic, whose principal neurotransmitters are epinephrine and norepinephrine; parasympathetic, whose principal neurotransmitter is acetylcholine; and nonadrenergic/noncholinergic, which include two subgroups—nitrergic, whose principal neurotransmitter is NO, and peptidergic, whose principal neurotransmitters are substance P, vasoactive intestinal peptide, calcitonin gene-related peptide, and ATP.
Each of these neurotransmitters acts through specific receptors on the vascular smooth-muscle cell to modulate intracellular calcium and, consequently, contractile tone. Norepinephrine activates α receptors, and epinephrine activates α and β receptors (adrenergic receptors); in most blood vessels, norepinephrine activates postjunctional α1 receptors in large arteries and α2 receptors in small arteries and arterioles, leading to vasoconstriction. Most blood vessels express β2-adrenergic receptors on their vascular smooth-muscle cells and respond to β agonists by cyclic AMP–dependent relaxation. Acetylcholine released from parasympathetic neurons binds to muscarinic receptors (of which there are five subtypes, M1–5) on vascular smooth-muscle cells to yield vasorelaxation. In addition, NO stimulates presynaptic neurons to release acetylcholine, which can stimulate the release of NO from the endothelium. Nitrergic neurons release NO produced by neuronal NO synthase, which causes vascular smooth-muscle cell relaxation via the cyclic GMP–dependent and –independent mechanisms described above. The peptidergic neurotransmitters all potently vasodilate, acting either directly or through endothelium-dependent NO release to decrease vascular smooth-muscle cell tone. For the detailed molecular physiology of the autonomic nervous system, see Chap. 454.
The endothelium modulates vascular smooth-muscle tone by the direct release of several effectors, including NO, prostacyclin, hydrogen sulfide, and endothelium-derived hyperpolarizing factor, all of which cause vasorelaxation, and endothelin, which causes vasoconstriction. The release of these endothelial effectors of vascular smooth-muscle cell tone is stimulated by mechanical (shear stress, cyclic strain, etc.) and biochemical mediators (purinergic agonists, muscarinic agonists, peptidergic agonists), with the biochemical mediators acting through endothelial receptors specific to each class. In addition to these local paracrine modulators of vascular smooth-muscle cell tone, circulating mediators can affect tone, including norepinephrine and epinephrine, vasopressin, angiotensin II, bradykinin, and the natriuretic peptides (atrial natriuretic peptide [ANP], brain natriuretic peptide [BNP], C-type natriuretic peptide [CNP], and dendroaspis natriuretic peptide [DNP]), as discussed above.
VASCULAR REGENERATION
Growth of new blood vessels can occur in response to conditions such as chronic hypoxemia and tissue ischemia. Growth factors, including vascular endothelial growth factor (VEGF) and forms of fibroblast growth factor (FGF), activate a signaling cascade that stimulates endothelial proliferation and tube formation, defined as angiogenesis. Guidance molecules, including members of the semaphorin family of secreted peptides, direct blood vessel patterning by attracting or repelling nascent endothelial tubes. The development of collateral vascular networks in the ischemic myocardium, an example of angiogenesis, can result from selective activation of endothelial progenitor cells, which may reside in the blood vessel wall or home to the ischemic tissue from the bone marrow. True arteriogenesis, or the development of a new blood vessel that includes all three cell layers, normally does not occur in the cardiovascular system of adult mammals. The molecular mechanisms and progenitor cells that can recapitulate blood vessel development de novo are under rapidly advancing study (Chaps. 88, 89e, and 90e).
VASCULAR PHARMACOGENOMICS
The last decade has witnessed considerable progress in efforts to define the genetic differences underlying individual variations in vascular pharmacologic responses. Many investigators have focused on receptors and enzymes associated with neurohumoral modulation of vascular function as well as hepatic enzymes that metabolize drugs that affect vascular tone. The genetic polymorphisms thus far associated with differences in vascular response often (but not invariably) relate to functional differences in the activity or expression of the receptor or enzyme of interest. Some of these polymorphisms appear to have different allele frequencies in specific ethnic groups.
CELLULAR BASIS OF CARDIAC CONTRACTION
CARDIAC ULTRASTRUCTURE
About three-fourths of the ventricular mass is composed of cardiomyocytes, normally 60–140 μm in length and 17–25 μm in diameter (Fig. 265e-4A). Each cell contains multiple, rodlike cross-banded strands (myofibrils) that run the length of the cell and are composed of serially repeating structures, the sarcomeres. The cytoplasm between the myofibrils contains other cell constituents, including the single centrally located nucleus, numerous mitochondria, and the intracellular membrane system, the sarcoplasmic reticulum.
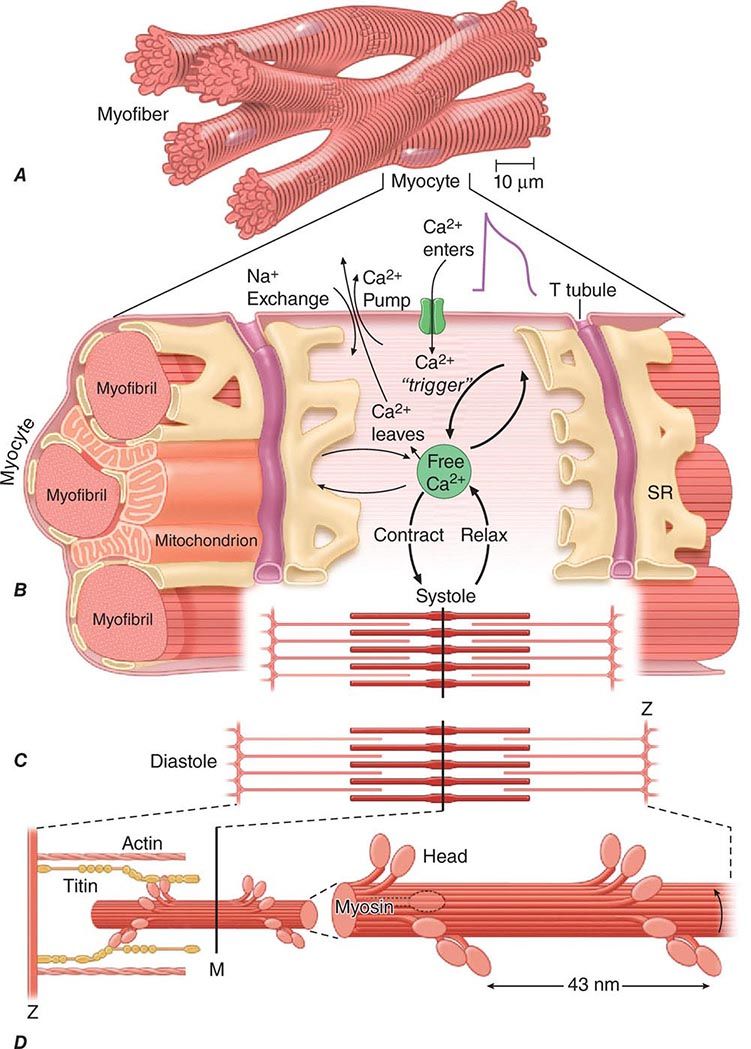
FIGURE 265e-4 A shows the branching myocytes making up the cardiac myofibers. B illustrates the critical role played by the changing [Ca2+] in the myocardial cytosol. Ca2+ ions are schematically shown as entering through the calcium channel that opens in response to the wave of depolarization that travels along the sarcolemma. These Ca2+ ions “trigger” the release of more calcium from the sarcoplasmic reticulum (SR) and thereby initiate a contraction-relaxation cycle. Eventually the small quantity of Ca2+ that has entered the cell leaves predominantly through an Na+/Ca2+ exchanger, with a lesser role for the sarcolemmal Ca2+ pump. The varying actin-myosin overlap is shown for (B) systole, when [Ca2+] is maximal, and (C) diastole, when [Ca2+] is minimal. D. The myosin heads, attached to the thick filaments, interact with the thin actin filaments. (From LH Opie: Heart Physiology: From Cell to Circulation, 4th ed. Philadelphia, Lippincott, Williams & Wilkins, 2004. Reprinted with permission. Copyright LH Opie, 2004.)
The sarcomere, the structural and functional unit of contraction, lies between adjacent Z lines, which are dark repeating bands that are apparent on transmission electron microscopy. The distance between Z lines varies with the degree of contraction or stretch of the muscle and ranges between 1.6 and 2.2 μm. Within the confines of the sarcomere are alternating light and dark bands, giving the myocardial fibers their striated appearance under the light microscope. At the center of the sarcomere is a dark band of constant length (1.5 μm), the A band, which is flanked by two lighter bands, the I bands, which are of variable length. The sarcomere of heart muscle, like that of skeletal muscle, consists of two sets of interdigitating myofilaments. Thicker filaments, composed principally of the protein myosin, traverse the A band; they are about 10 nm (100 Å) in diameter, with tapered ends. Thinner filaments, composed primarily of actin, course from the Z lines through the I band into the A band; they are approximately 5 nm (50 Å) in diameter and 1.0 μm in length. Thus, thick and thin filaments overlap only within the (dark) A band, whereas the (light) I band contains only thin filaments. On electron-microscopic examination, bridges may be seen to extend between the thick and thin filaments within the A band; these are myosin heads (see below) bound to actin filaments.
THE CONTRACTILE PROCESS
The sliding filament model for muscle contraction rests on the fundamental observation that both the thick and the thin filaments are constant in overall length during both contraction and relaxation. With activation, the actin filaments are propelled farther into the A band. In the process, the A band remains constant in length, whereas the I band shortens and the Z lines move toward one another.
The myosin molecule is a complex, asymmetric fibrous protein with a molecular mass of about 500,000 Da; it has a rodlike portion that is about 150 nm (1500 Å) in length with a globular portion (head) at its end. These globular portions of myosin form the bridges between the myosin and actin molecules and are the site of ATPase activity. In forming the thick myofilament, which is composed of ~300 longitudinally stacked myosin molecules, the rodlike segments of the myosin molecules are laid down in an orderly, polarized manner, leaving the globular portions projecting outward so that they can interact with actin to generate force and shortening (Fig. 265e-4B).
Actin has a molecular mass of about 47,000 Da. The thin filament consists of a double helix of two chains of actin molecules wound about each other on a larger molecule, tropomyosin. A group of regulatory proteins—troponins C, I, and T—are spaced at regular intervals on this filament (Fig. 265e-5). In contrast to myosin, actin lacks intrinsic enzymatic activity but does combine reversibly with myosin in the presence of ATP and Ca2+. The calcium ion activates the myosin ATPase, which in turn breaks down ATP, the energy source for contraction (Fig. 265e-5). The activity of myosin ATPase determines the rate of forming and breaking of the actomyosin cross-bridges and ultimately the velocity of muscle contraction. In relaxed muscle, tropomyosin inhibits this interaction. Titin (Fig. 265e-4D) is a large, flexible, myofibrillar protein that connects myosin to the Z line; its stretching contributes to the elasticity of the heart. Dystrophin is a long cytoskeletal protein that has an amino-terminal actin-binding domain and a carboxy-terminal domain that binds to the dystroglycan complex at adherens junctions on the cell membrane, thus tethering the sarcomere to the cell membrane at regions tightly coupled to adjacent contracting myocytes. Mutations in components of the dystrophin complex lead to muscular dystrophy and associated cardiomyopathy.
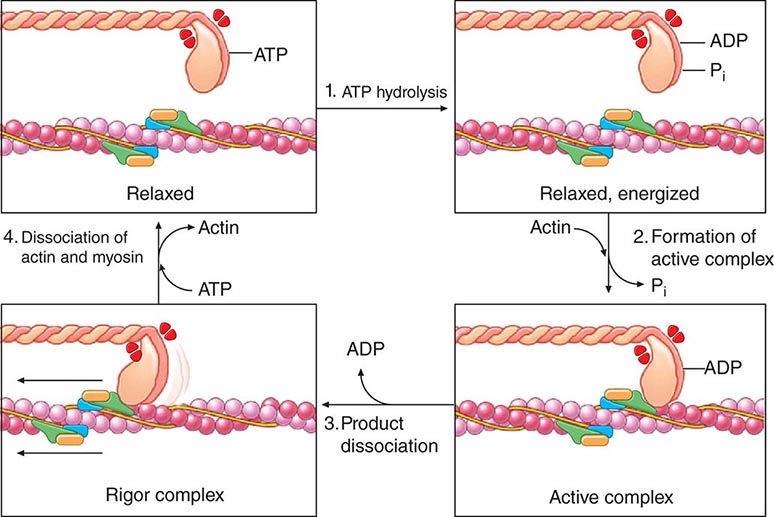
FIGURE 265e-5 Four steps in cardiac muscle contraction and relaxation. In relaxed muscle (upper left), ATP bound to the myosin cross-bridge dissociates the thick and thin filaments. Step 1: Hydrolysis of myosin-bound ATP by the ATPase site on the myosin head transfers the chemical energy of the nucleotide to the activated cross-bridge (upper right). When cytosolic Ca2+ concentration is low, as in relaxed muscle, the reaction cannot proceed because tropomyosin and the troponin complex on the thin filament do not allow the active sites on actin to interact with the cross-bridges. Therefore, even though the cross-bridges are energized, they cannot interact with actin. Step 2: When Ca2+ binding to troponin C has exposed active sites on the thin filament, actin interacts with the myosin cross-bridges to form an active complex (lower right) in which the energy derived from ATP is retained in the actin-bound cross-bridge, whose orientation has not yet shifted. Step 3: The muscle contracts when ADP dissociates from the cross-bridge. This step leads to the formation of the low-energy rigor complex (lower left) in which the chemical energy derived from ATP hydrolysis has been expended to perform mechanical work (the “rowing” motion of the cross-bridge). Step 4: The muscle returns to its resting state, and the cycle ends when a new molecule of ATP binds to the rigor complex and dissociates the cross-bridge from the thin filament. This cycle continues until calcium is dissociated from troponin C in the thin filament, which causes the contractile proteins to return to the resting state with the cross-bridge in the energized state. ADP, adenosine diphosphate; ATP, adenosine triphosphate; ATPase, adenosine triphosphatase. (From AM Katz: Heart failure: Cardiac function and dysfunction, in Atlas of Heart Diseases, 3rd ed, WS Colucci [ed]. Philadelphia, Current Medicine, 2002. Reprinted with permission.)
During activation of the cardiac myocyte, Ca2+ becomes attached to one of three components of the heterotrimer troponin C, which results in a conformational change in the regulatory protein tropomyosin; the latter, in turn, exposes the actin cross-bridge interaction sites (Fig. 265e-5). Repetitive interaction between myosin heads and actin filaments is termed cross-bridge cycling, which results in sliding of the actin along the myosin filaments, ultimately causing muscle shortening and/or the development of tension. The splitting of ATP then dissociates the myosin cross-bridge from actin. In the presence of ATP (Fig. 265e-5), linkages between actin and myosin filaments are made and broken cyclically as long as sufficient Ca2+ is present; these linkages cease when [Ca2+] falls below a critical level, and the troponin-tropomyosin complex once more prevents interactions between the myosin cross-bridges and actin filaments (Fig. 265e-6).
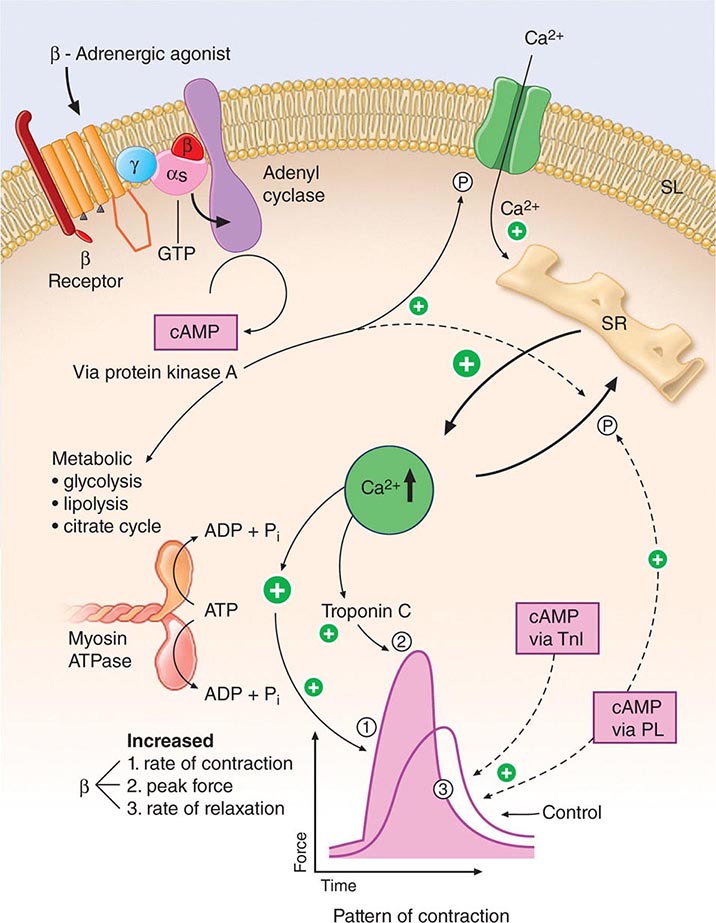
FIGURE 265e-6 Signal systems involved in positive inotropic and lusitropic (enhanced relaxation) effects of β-adrenergic stimulation. When the β-adrenergic agonist interacts with the β receptor, a series of G protein–mediated changes leads to activation of adenylyl cyclase and the formation of cyclic adenosine monophosphate (cAMP). The latter acts via protein kinase A to stimulate metabolism (left) and phosphorylate the Ca2+ channel protein (right). The result is an enhanced opening probability of the Ca2+ channel, thereby increasing the inward movement of Ca2+ ions through the sarcolemma (SL) of the T tubule. These Ca2+ ions release more calcium from the sarcoplasmic reticulum (SR) to increase cytosolic Ca2+ and activate troponin C. Ca2+ ions also increase the rate of breakdown of adenosine triphosphate (ATP) to adenosine diphosphate (ADP) and inorganic phosphate (Pi). Enhanced myosin ATPase activity explains the increased rate of contraction, with increased activation of troponin C explaining increased peak force development. An increased rate of relaxation results from the ability of cAMP to activate as well the protein phospholamban, situated on the membrane of the SR, that controls the rate of uptake of calcium into the SR. The latter effect explains enhanced relaxation (lusitropic effect). P, phosphorylation; PL, phospholamban; TnI, troponin I. (Modified from LH Opie: Heart Physiology: From Cell to Circulation, 4th ed. Philadelphia, Lippincott, Williams & Wilkins, 2004. Reprinted with permission. Copyright LH Opie, 2004.)
Intracytoplasmic Ca2+ is a principal determinant of the inotropic state of the heart. Most agents that stimulate myocardial contractility (positive inotropic stimuli), including the digitalis glycosides and β-adrenergic agonists, increase the [Ca2+] in the vicinity of the myofilaments, which in turn triggers cross-bridge cycling. Increased impulse traffic in the cardiac adrenergic nerves stimulates myocardial contractility as a consequence of the release of norepinephrine from cardiac adrenergic nerve endings. Norepinephrine activates myocardial β receptors and, through the Gs-stimulated guanine nucleotide-binding protein, activates the enzyme adenylyl cyclase, which leads to the formation of the intracellular second messenger cyclic AMP from ATP (Fig. 265e-6). Cyclic AMP in turn activates protein kinase A (PKA), which phosphorylates the Ca2+ channel in the myocardial sarcolemma, thereby enhancing the influx of Ca2+ into the myocyte. Other functions of PKA are discussed below.
The sarcoplasmic reticulum (SR) (Fig. 265e-7), a complex network of anastomosing intracellular channels, invests the myofibrils. Its longitudinally disposed tubules closely invest the surfaces of individual sarcomeres but have no direct continuity with the outside of the cell. However, closely related to the SR, both structurally and functionally, are the transverse tubules, or T system, formed by tubelike invaginations of the sarcolemma that extend into the myocardial fiber along the Z lines, i.e., the ends of the sarcomeres.
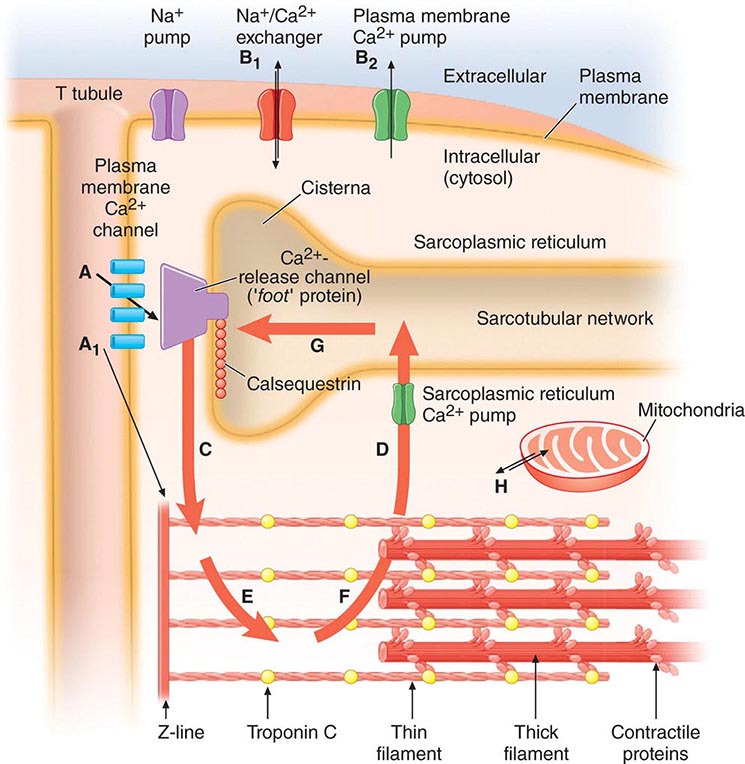
FIGURE 265e-7 The Ca2+ fluxes and key structures involved in cardiac excitation-contraction coupling. The arrows denote the direction of Ca2+ fluxes. The thickness of each arrow indicates the magnitude of the calcium flux. Two Ca2+ cycles regulate excitation-contraction coupling and relaxation. The larger cycle is entirely intracellular and involves Ca2+ fluxes into and out of the sarcoplasmic reticulum, as well as Ca2+ binding to and release from troponin C. The smaller extracellular Ca2+ cycle occurs when this cation moves into and out of the cell. The action potential opens plasma membrane Ca2+ channels to allow passive entry of Ca2+ into the cell from the extracellular fluid (arrow A). Only a small portion of the Ca2+ that enters the cell directly activates the contractile proteins (arrow A1). The extracellular cycle is completed when Ca2+ is actively transported back out to the extracellular fluid by way of two plasma membrane fluxes mediated by the sodium-calcium exchanger (arrow B1) and the plasma membrane calcium pump (arrow B2). In the intracellular Ca2+ cycle, passive Ca2+ release occurs through channels in the cisternae (arrow C) and initiates contraction; active Ca2+ uptake by the Ca2+ pump of the sarcotubular network (arrow D) relaxes the heart. Diffusion of Ca2+ within the sarcoplasmic reticulum (arrow G) returns this activator cation to the cisternae, where it is stored in a complex with calsequestrin and other calcium-binding proteins. Ca2+ released from the sarcoplasmic reticulum initiates systole when it binds to troponin C (arrow E). Lowering of cytosolic [Ca2+] by the sarcoplasmic reticulum (SR) causes this ion to dissociate from troponin (arrow F) and relaxes the heart. Ca2+ also may move between mitochondria and cytoplasm (H). (Adapted from AM Katz: Physiology of the Heart, 4th ed. Philadelphia, Lippincott, Williams & Wilkins, 2005, with permission.)
CARDIAC ACTIVATION
In the inactive state, the cardiac cell is electrically polarized; i.e., the interior has a negative charge relative to the outside of the cell, with a transmembrane potential of –80 to –100 mV (Chap. 273e). The sarcolemma, which in the resting state is largely impermeable to Na+, has a Na+– and K+-stimulating pump energized by ATP that extrudes Na+ from the cell; this pump plays a critical role in establishing the resting potential. Thus, intracellular [K+] is relatively high and [Na+] is far lower; conversely, extracellular [Na+] is high and [K+] is low. At the same time, in the resting state, extracellular [Ca2+] greatly exceeds free intracellular [Ca2+].
The action potential has four phases (see Fig. 273e-1B). During the plateau of the action potential (phase 2), there is a slow inward current through L-type Ca2+ channels in the sarcolemma (Fig. 265e-7). The depolarizing current not only extends across the surface of the cell but penetrates deeply into the cell by way of the ramifying T tubular system. The absolute quantity of Ca2+ that crosses the sarcolemma and the T system is relatively small and by itself appears to be insufficient to bring about full activation of the contractile apparatus. However, this Ca2+ current triggers the release of much larger quantities of Ca2+ from the SR, a process termed Ca2+-induced Ca2+ release. The latter is a major determinant of intracytoplasmic [Ca2+] and therefore of myocardial contractility.
Ca2+ is released from the SR through a Ca2+ release channel, a cardiac isoform of the ryanodine receptor (RyR2), which controls intracytoplasmic [Ca2+] and, as in vascular smooth-muscle cells, leads to the local changes in intracellular [Ca2+] called calcium sparks. A number of regulatory proteins, including calstabin 2, inhibit RyR2 and thereby the release of Ca2+ from the SR. PKA dissociates calstabin from the RyR2, enhancing Ca2+ release and thereby myocardial contractility. Excessive plasma catecholamine levels and cardiac sympathetic neuronal release of norepinephrine cause hyperphosphorylation of PKA, leading to calstabin 2–depleted RyR2. The latter depletes SR Ca2+ stores and thereby impairs cardiac contraction, leading to heart failure, and also triggers ventricular arrhythmias.
The Ca2+ released from the SR then diffuses toward the myofibrils, where, as already described, it combines with troponin C (Fig. 265e-6). By repressing this inhibitor of contraction, Ca2+ activates the myofilaments to shorten. During repolarization, the activity of the Ca2+ pump in the SR, the SR Ca2+ ATPase (SERCA2A), reaccumulates Ca2+ against a concentration gradient, and the Ca2+ is stored in the SR by its attachment to a protein, calsequestrin. This reaccumulation of Ca2+ is an energy (ATP)-requiring process that lowers the cytoplasmic [Ca2+] to a level that inhibits the actomyosin interaction responsible for contraction, and in this manner leads to myocardial relaxation. Also, there is an exchange of Ca2+ for Na+ at the sarcolemma (Fig. 265e-7), reducing the cytoplasmic [Ca2+]. Cyclic AMP–dependent PKA phosphorylates the SR protein phospholamban; the latter, in turn, permits activation of the Ca2+ pump, thereby increasing the uptake of Ca2+ by the SR, accelerating the rate of relaxation, and providing larger quantities of Ca2+ in the SR for release by subsequent depolarization, thereby stimulating contraction.
Thus, the combination of the cell membrane, transverse tubules, and SR, with their ability to transmit the action potential and release and then reaccumulate Ca2+, plays a fundamental role in the rhythmic contraction and relaxation of heart muscle. Genetic or pharmacologic alterations of any component, whatever its etiology, can disturb these functions.
CONTROL OF CARDIAC PERFORMANCE AND OUTPUT
The extent of shortening of heart muscle and, therefore, the stroke volume of the ventricle in the intact heart depend on three major influences: (1) the length of the muscle at the onset of contraction, i.e., the preload; (2) the tension that the muscle is called on to develop during contraction, i.e., the afterload; and (3) the contractility of the muscle, i.e., the extent and velocity of shortening at any given preload and afterload. The major determinants of preload, afterload, and contractility are shown in Table 265e-2.
DETERMINANTS OF STROKE VOLUME |
aArrows indicate directional effects of determinants of contractility. bContractility rises initially but later becomes depressed.
THE ROLE OF MUSCLE LENGTH (PRELOAD)
The preload determines the length of the sarcomeres at the onset of contraction. The length of the sarcomeres associated with the most forceful contraction is ~2.2 μm. This length provides the optimum configuration for the interaction between the two sets of myofilaments. The length of the sarcomere also regulates the extent of activation of the contractile system, i.e., its sensitivity to Ca2+. According to this concept, termed length-dependent activation, myofilament sensitivity to Ca2+ is also maximal at the optimal sarcomere length. The relation between the initial length of the muscle fibers and the developed force has prime importance for the function of heart muscle. This relationship forms the basis of Starling’s law of the heart, which states that within limits, the force of ventricular contraction depends on the end-diastolic length of the cardiac muscle; in the intact heart, the latter relates closely to the ventricular end-diastolic volume.
CARDIAC PERFORMANCE
The ventricular end-diastolic or “filling” pressure sometimes is used as a surrogate for the end-diastolic volume. In isolated heart and heart-lung preparations, the stroke volume varies directly with the end-diastolic fiber length (preload) and inversely with the arterial resistance (afterload), and as the heart fails—i.e., as its contractility declines—it delivers a progressively smaller stroke volume from a normal or even elevated end-diastolic volume. The relation between the ventricular end-diastolic pressure and the stroke work of the ventricle (the ventricular function curve) provides a useful definition of the level of contractility of the heart in the intact organism. An increase in contractility is accompanied by a shift of the ventricular function curve upward and to the left (greater stroke work at any level of ventricular end-diastolic pressure, or lower end-diastolic volume at any level of stroke work), whereas a shift downward and to the right characterizes depression of contractility (Fig. 265e-8).
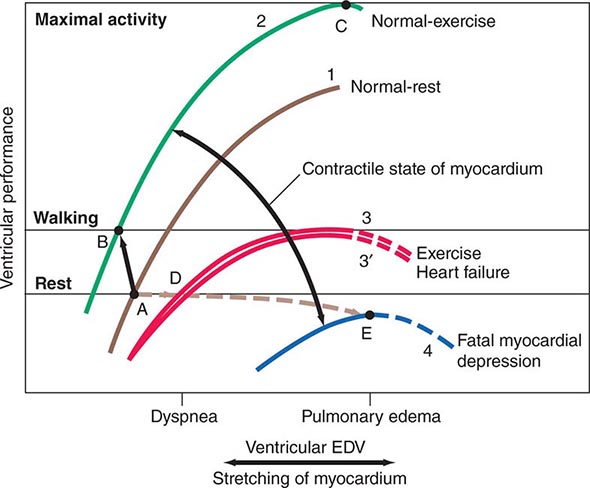
FIGURE 265e-8 The interrelations among influences on ventricular end-diastolic volume (EDV) through stretching of the myocardium and the contractile state of the myocardium. Levels of ventricular EDV associated with filling pressures that result in dyspnea and pulmonary edema are shown on the abscissa. Levels of ventricular performance required when the subject is at rest, while walking, and during maximal activity are designated on the ordinate. The broken lines are the descending limbs of the ventricular-performance curves, which are rarely seen during life but show the level of ventricular performance if end-diastolic volume could be elevated to very high levels. For further explanation, see text. (Modified from WS Colucci and E Braunwald: Pathophysiology of heart failure, in Braunwald’s Heart Disease, 7th ed, DP Zipes et al [eds]. Philadelphia: Elsevier, 2005, pp 509–538.)
VENTRICULAR AFTERLOAD
In the intact heart, as in isolated cardiac muscle, the extent and velocity of shortening of ventricular muscle fibers at any level of preload and of myocardial contractility relate inversely to the afterload, i.e., the load that opposes shortening. In the intact heart, the afterload may be defined as the tension developed in the ventricular wall during ejection. Afterload is determined by the aortic pressure as well as by the volume and thickness of the ventricular cavity. Laplace’s law states that the tension of the myocardial fiber is the product of the intracavitary ventricular pressure and ventricular radius divided by wall thickness. Therefore, at any particular level of aortic pressure, the afterload on a dilated left ventricle exceeds that on a normal-sized ventricle. Conversely, at the same aortic pressure and ventricular diastolic volume, the afterload on a hypertrophied ventricle is lower than of a normal chamber. The aortic pressure in turn depends on the peripheral vascular resistance, the physical characteristics of the arterial tree, and the volume of blood it contains at the onset of ejection.
Ventricular afterload critically regulates cardiovascular performance (Fig. 265e-9). As already noted, elevations in both preload and contractility increase myocardial fiber shortening, whereas increases in afterload reduce it. The extent of myocardial fiber shortening and left ventricular size determine stroke volume. An increase in arterial pressure induced by vasoconstriction, for example, augments afterload, which opposes myocardial fiber shortening, reducing stroke volume.
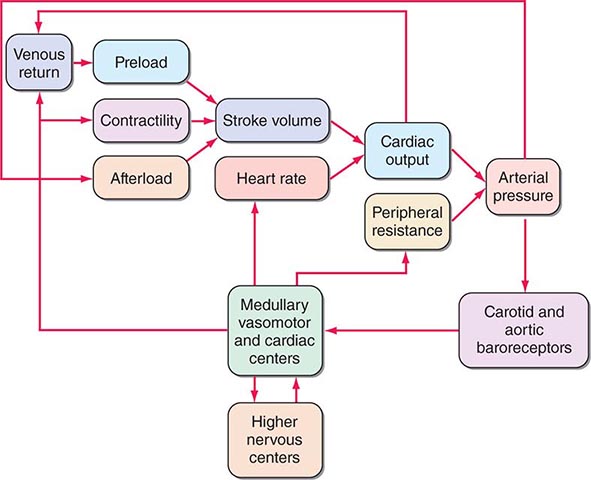
FIGURE 265e-9 Interactions in the intact circulation of preload, contractility, and afterload in producing stroke volume. Stroke volume combined with heart rate determines cardiac output, which, when combined with peripheral vascular resistance, determines arterial pressure for tissue perfusion. The characteristics of the arterial system also contribute to afterload, an increase that reduces stroke volume. The interaction of these components with carotid and aortic arch baroreceptors provides a feedback mechanism to higher medullary and vasomotor cardiac centers and to higher levels in the central nervous system to effect a modulating influence on heart rate, peripheral vascular resistance, venous return, and contractility. (From MR Starling: Physiology of myocardial contraction, in Atlas of Heart Failure: Cardiac Function and Dysfunction, 3rd ed, WS Colucci and E Braunwald [eds]. Philadelphia: Current Medicine, 2002, pp 19–35.)
When myocardial contractility becomes impaired and the ventricle dilates, afterload rises (Laplace’s law) and limits cardiac output. Increased afterload also may result from neural and humoral stimuli that occur in response to a fall in cardiac output. This increased afterload may reduce cardiac output further, thereby increasing ventricular volume and initiating a vicious circle, especially in patients with ischemic heart disease and limited myocardial O2 supply. Treatment with vasodilators has the opposite effect; when afterload is reduced, cardiac output rises (Chap. 279).
Under normal circumstances, the various influences acting on cardiac performance enumerated above interact in a complex fashion to maintain cardiac output at a level appropriate to the requirements of the metabolizing tissues (Fig. 265e-9); interference with a single mechanism may not influence the cardiac output. For example, a moderate reduction of blood volume or the loss of the atrial contribution to ventricular contraction ordinarily can be sustained without a reduction in the cardiac output at rest. Under these circumstances, other factors, such as increases in the frequency of adrenergic nerve impulses to the heart, heart rate, and venous tone, will serve as compensatory mechanisms and sustain cardiac output in a normal individual.
EXERCISE
The integrated response to exercise illustrates the interactions among the three determinants of stroke volume: preload, afterload, and contractility (Fig. 265e-8). Hyperventilation, the pumping action of the exercising muscles, and venoconstriction during exercise all augment venous return and hence ventricular filling and preload (Table 265e-2). Simultaneously, the increase in the adrenergic nerve impulse traffic to the myocardium, the increased concentration of circulating catecholamines, and the tachycardia that occur during exercise combine to augment the contractility of the myocardium (Fig. 265e-8, curves 1 and 2) and together elevate stroke volume and stroke work, without a change in or even a reduction of end-diastolic pressure and volume (Fig. 265e-8, points A and B). Vasodilation occurs in the exercising muscles, thus tending to limit the increase in arterial pressure that otherwise would occur as cardiac output rises to levels as high as five times greater than basal levels during maximal exercise. This vasodilation ultimately allows the achievement of a greatly elevated cardiac output during exercise at an arterial pressure only moderately higher than in the resting state.
ASSESSMENT OF CARDIAC FUNCTION
Several techniques can define impaired cardiac function in clinical practice. The cardiac output and stroke volume may be depressed in the presence of heart failure, but not uncommonly, these variables are within normal limits in this condition. A somewhat more sensitive index of cardiac function is the ejection fraction, i.e., the ratio of stroke volume to end-diastolic volume (normal value = 67 ± 8%), which is frequently depressed in systolic heart failure even when the stroke volume itself is normal. Alternatively, abnormally elevated ventricular end-diastolic volume (normal value = 75 ± 20 mL/m2) or end-systolic volume (normal value = 25 ± 7 mL/m2) signifies impairment of left ventricular systolic function.
Noninvasive techniques, particularly echocardiography as well as radionuclide scintigraphy and cardiac magnetic resonance imaging (MRI) (Chap. 270e), have great value in the clinical assessment of myocardial function. They provide measurements of end-diastolic and end-systolic volumes, ejection fraction, and systolic shortening rate, and they allow assessment of ventricular filling (see below) as well as regional contraction and relaxation. The latter measurements are particularly important in ischemic heart disease, as myocardial infarction causes regional myocardial damage.
A limitation of measurements of cardiac output, ejection fraction, and ventricular volumes in assessing cardiac function is that ventricular loading conditions strongly influence these variables. Thus, a depressed ejection fraction and lowered cardiac output may occur in patients with normal ventricular function but reduced preload, as occurs in hypovolemia, or with increased afterload, as occurs in acutely elevated arterial pressure.
The end-systolic left ventricular pressure-volume relationship is a particularly useful index of ventricular performance because it does not depend on preload and afterload (Fig. 265e-10). At any level of myocardial contractility, left ventricular end-systolic volume varies inversely with end-systolic pressure; as contractility declines, end-systolic volume (at any level of end-systolic pressure) rises.
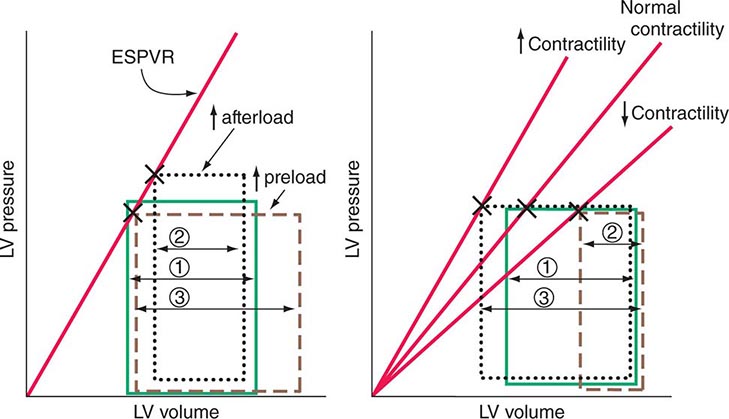
FIGURE 265e-10 The responses of the left ventricle to increased afterload, increased preload, and increased and reduced contractility are shown in the pressure-volume plane. Left. Effects of increases in preload and afterload on the pressure-volume loop. Because there has been no change in contractility, the end-systolic pressure-volume relationship (ESPVR) is unchanged. With an increase in afterload, stroke volume falls (1 → 2); with an increase in preload, stroke volume rises (1 → 3). Right. With increased myocardial contractility and constant left ventricular end-diastolic volume, the ESPVR moves to the left of the normal line (lower end-systolic volume at any end-systolic pressure) and stroke volume rises (1 → 3). With reduced myocardial contractility, the ESPVR moves to the right; end-systolic volume is increased, and stroke volume falls (1 → 2).
DIASTOLIC FUNCTION
Ventricular filling is influenced by the extent and speed of myocardial relaxation, which in turn depends on the rate of uptake of Ca2+ by the SR; the latter may be enhanced by adrenergic activation and reduced by ischemia, which reduces the ATP available for pumping Ca2+ into the SR (see above). The stiffness of the ventricular wall also may impede filling. Ventricular stiffness increases with hypertrophy and conditions that infiltrate the ventricle, such as amyloid, or is caused by an extrinsic constraint (e.g., pericardial compression) (Fig. 265e-11).
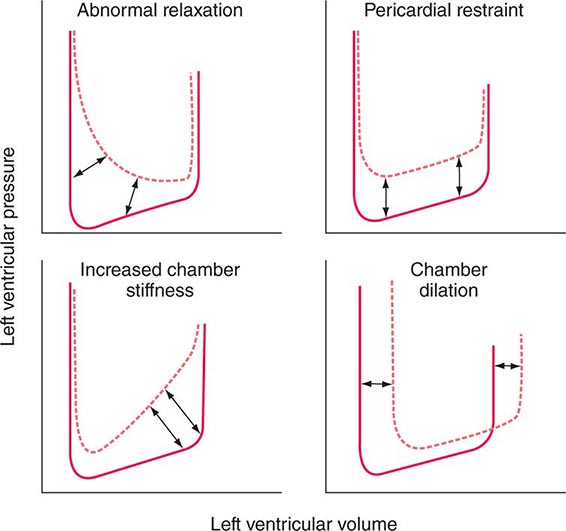
FIGURE 265e-11 Mechanisms that cause diastolic dysfunction reflected in the pressure-volume relation. The bottom half of the pressure-volume loop is depicted. Solid lines represent normal subjects; broken lines represent patients with diastolic dysfunction. (From JD Carroll et al: The differential effects of positive inotropic and vasodilator therapy on diastolic properties in patients with congestive cardiomyopathy. Circulation 74:815, 1986; with permission.)
Ventricular filling can be assessed by continuously measuring the velocity of flow across the mitral valve using Doppler ultrasound. Normally, the velocity of inflow is more rapid in early diastole than during atrial systole; with mild to moderately impaired relaxation, the rate of early diastolic filling declines, whereas the rate of presystolic filling rises. With further impairment of filling, the pattern is “pseudonormalized,” and early ventricular filling becomes more rapid as left atrial pressure upstream to the stiff left ventricle rises.
CARDIAC METABOLISM
The heart requires a continuous supply of energy (in the form of ATP) not only to perform its mechanical pumping functions, but also to regulate intracellular and transsarcolemmal ionic movements and concentration gradients. Among its pumping functions, the development of tension, the frequency of contraction, and the level of myocardial contractility are the principal determinants of the heart’s substantial energy needs, making its O2 requirements approximately 15% of that of the entire organism.
Most ATP production depends on the oxidation of substrate (glucose and free fatty acids [FFAs]). Myocardial FFAs are derived from circulating FFAs, which result principally from lipolysis in adipose tissue, whereas the myocyte’s glucose derives from plasma as well as from the cell’s breakdown of its glycogen stores (glycogenolysis). These two principal sources of acetyl coenzyme A in cardiac muscle vary reciprocally. Glucose is broken down in the cytoplasm into a three-carbon product, pyruvate, which passes into the mitochondria, where it is metabolized to the two-carbon fragment, acetyl-CoA, and undergoes oxidation. FFAs are converted to acyl-CoA in the cytoplasm and acetyl-CoA in the mitochondria. Acetyl-CoA enters the citric acid (Krebs) cycle to produce ATP by oxidative phosphorylation within the mitochondria; ATP then enters the cytoplasm from the mitochondrial compartment. Intracellular ADP, resulting from the breakdown of ATP, enhances mitochondrial ATP production.
In the fasted, resting state, circulating FFA concentrations and their myocardial uptake are high, and they furnish most of the heart’s acetyl-CoA (~70%). In the fed state, with elevations of blood glucose and insulin, glucose oxidation increases and FFA oxidation subsides. Increased cardiac work, the administration of inotropic agents, hypoxia, and mild ischemia all enhance myocardial glucose uptake, glucose production resulting from glycogenolysis, and glucose metabolism to pyruvate (glycolysis). By contrast, β-adrenergic stimulation, as occurs during stress, raises the circulating levels and metabolism of FFAs in favor of glucose. Severe ischemia inhibits the cytoplasmic enzyme pyruvate dehydrogenase, and despite both glycogen and glucose breakdown, glucose is metabolized only to lactic acid (anaerobic glycolysis), which does not enter the citric acid cycle. Anaerobic glycolysis produces much less ATP than does aerobic glucose metabolism, in which glucose is metabolized to pyruvate and subsequently oxidized to CO2. High concentrations of circulating FFAs, which can occur when adrenergic stimulation is superimposed on severe ischemia, reduce oxidative phosphorylation and also cause ATP wastage; the myocardial content of ATP declines and impairs myocardial contraction. In addition, products of FFA breakdown can exert toxic effects on cardiac cell membranes and may be arrhythmogenic.
Myocardial energy is stored as creatine phosphate (CP), which is in equilibrium with ATP, the immediate source of energy. In states of reduced energy availability, the CP stores decline first. Cardiac hypertrophy, fibrosis, tachycardia, increased wall tension resulting from ventricular dilation, and increased intracytoplasmic [Ca2+] all contribute to increased myocardial energy needs. When coupled with reduced coronary flow reserve, as occurs with obstruction of coronary arteries or abnormalities of the coronary microcirculation, an imbalance in myocardial ATP production relative to demand may occur, and the resulting ischemia can worsen or cause heart failure.
Developmental Biology of the Cardiovascular System The heart is the first organ to form during embryogenesis (Fig. 265e-12) and must accomplish the simultaneous challenges of circulating blood, nutrients, and oxygen to the other forming organs while continuing to grow and undergo complex morphogenetic changes. Early progenitors of the heart arise within very early crescent-shaped fields of lateral splanchnic mesoderm under the influence of multiple signals, including those derived from neural ectoderm long before neural tube closure. Early cardiac precursors express genes encoding regulatory transcription factors that play reiterated roles in cardiac development, such as NKX2-5 and GATA4. Mutations in these genes are responsible for some forms of inherited congenital heart disease. Cardiac precursors coalesce to form a midline heart tube composed of a single cell layer of endocardium surrounded by a single layer of myocardial precursors. The caudal, inflow region of the heart tube, which is destined to adopt a more rostral final position, represents the atrial anlagen, whereas the rostral, outflow portion of the tube forms the truncus arteriosus, which divides to produce the aorta and the proximal pulmonary artery. Between these extremes lie the structural precursors of the ventricles.
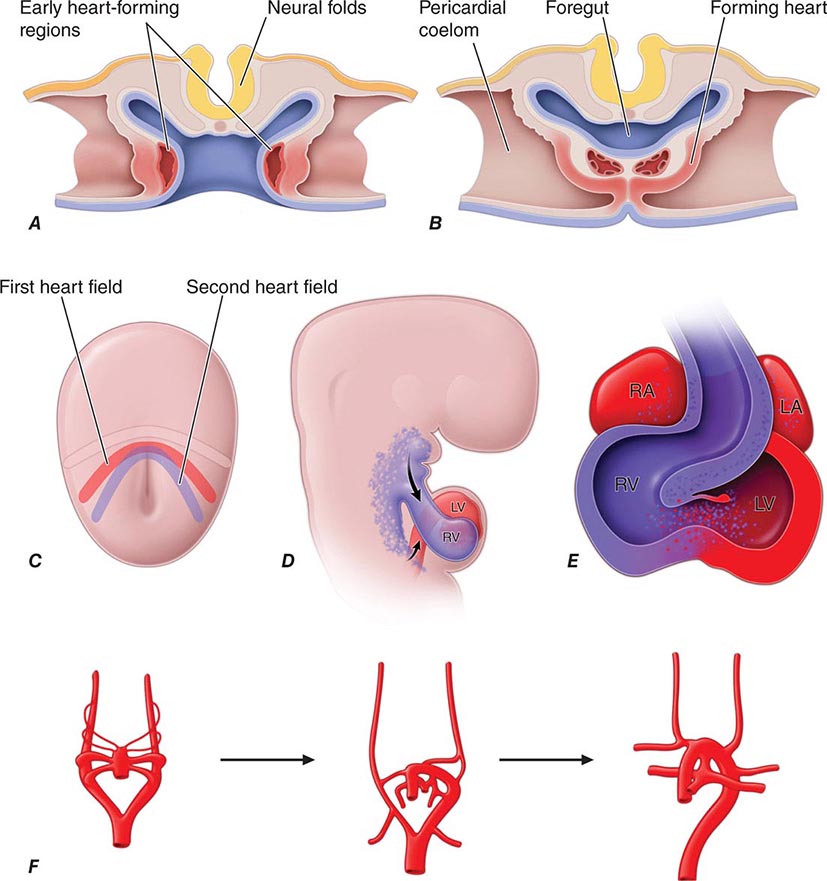
FIGURE 265e-12 A. Schematic depiction of a transverse section through an early embryo depicts the bilateral regions where early heart tubes form. B. The bilateral heart tubes subsequently migrate to the midline and fuse to form the linear heart tube. C. At the early cardiac crescent stage of embryonic development, cardiac precursors include a primary heart field fated to form the linear heart tube and a second heart field fated to add myocardium to the inflow and outflow poles of the heart. D. Second heart field cells populate the pharyngeal region before subsequently migrating to the maturing heart. E. Large portions of the right ventricle and outflow tract and some cells within the atria derive from the second heart field. F. The aortic arch arteries form as symmetric sets of vessels that then remodel under the influence of the neural crest to form the asymmetric mature vasculature. LA, left atrium; LV, left ventricle; RA, right atrium; RV, right ventricle.
The linear heart tube undergoes an asymmetric looping process (the first gross evidence of left-right asymmetry in the developing embryo), which positions the portion of the heart tube destined to become the left ventricle to the left of the more rostral precursors of the right ventricle and outflow tract. Looping is coordinated with chamber specification and ballooning of various regions of the heart tube to produce the presumptive atria and ventricles.
Relatively recent work has demonstrated that significant portions of the right ventricle are formed by cells that are added to the developing heart after looping has occurred. These cells, which are derived from what is called the second heart field, migrate to the heart from the ventral pharynx and express markers that allow for their identification, including Islet-1. Different embryologic origins of cells within the right and left ventricles may help explain why some forms of congenital and adult heart diseases affect these regions of the heart to varying degrees.
After looping and chamber formation, a series of septation events divide the left and right sides of the heart, separate the atria from the ventricles, and form the aorta and pulmonary artery from the truncus arteriosus. Cardiac valves form between the atria and the ventricles and between the ventricles and the outflow vessels. Early in development, the single layer of myocardial cells secretes an extracellular matrix rich in hyaluronic acid. This extracellular matrix, termed “cardiac jelly,” accumulates within the endocardial cushions, precursors of the cardiac valves. Signals from overlying myocardial cells, including members of the transforming growth factor β family, trigger migration, invasion, and phenotypic changes of underlying endocardial cells, which undergo an epithelial-mesenchymal transformation and invade the cardiac jelly to cellularize the endocardial cushions. Mesenchymal components proliferate and remodel to form the mature valve leaflets.
The great vessels form as a series of bilaterally symmetric aortic arch arteries that undergo asymmetric remodeling events to form the mature vasculature. The immigration of neural crest cells that arise in the dorsal neural tube orchestrates this process. These cells are required for aortic arch remodeling and septation of the truncus arteriosus. They develop into smooth-muscle cells within the tunica media of the aortic arch, the ductus arteriosus, and the carotid arteries. Smooth-muscle cells within the descending aorta arise from a different embryologic source, the lateral plate mesoderm, and smooth muscle of the proximal outflow tract arises from the second heart field. Neural crest cells are sensitive to both vitamin A and folic acid, and congenital heart disease involving abnormal remodeling of the aortic arch arteries has been associated with maternal deficiencies of these vitamins. Congenital heart disease involving the outflow tract can be associated with other defects of neural crest, such as cleft palate or craniofacial abnormalities.
Coronary artery formation requires yet another cell population that initiates extrinsic to the embryonic heart fields. Epicardial cells arise in the proepicardial organ, a derivative of the septum transversum, which also contributes to the fibrous portion of the diaphragm and to the liver. Proepicardial cells contribute to the smooth-muscle cells of the coronary arteries and are required for their proper patterning. Other cell types within the heart, including fibroblasts and potentially some myocardial and endocardial cells, also can arise from the proepicardium.
The cardiac conduction system, which functions both to generate and to propagate electrical impulses, develops primarily from multipotential cardiac precursors, which also give rise to cardiac muscle. The conduction system is composed of slow-conducting (proximal) components, such as the sinoatrial (SA) and atrioventricular (AV) nodes, as well as fast-conducting (distal) components, including the His bundle, bundle branches, and Purkinje fibers. The AV node primarily serves to delay the electrical impulse between atria and ventricles (manifesting decremental conduction), whereas the distal conduction system rapidly delivers the impulse throughout the ventricles. Significant recent attention has been focused on the embryologic origins of various components of the specialized conduction network. Precursors within the sinus venosus give rise to the SA node, whereas those within the AV canal mature into heterogeneous cell types that compose the AV node. Myocardial cells transdifferentiate into Purkinje fibers to form the distal conduction system. Fast and slow conducting cell types within the nodes and bundles are characterized by expression of distinct gap junction proteins, including connexins, and ion channels that characterize unique cell fates and electrical properties of the tissues. Developmental defects in conduction system morphogenesis and lineage determination can lead to various electrophysiologic disorders, including congenital heart block and preexcitation syndromes such as the Wolff-Parkinson-White syndrome (Chap. 276).
Studies of cardiac stem and progenitor cells suggest that progressive lineage restriction results in the gradual and stepwise determination of mature cell fates within the heart, with early precursors capable of adopting endothelial, smooth-muscle, or cardiac phenotypes, and subsequent further specialization into atrial, ventricular, and specialized conduction cell types.
REGENERATING CARDIAC TISSUE
Until very recently, adult mammalian myocardial cells were viewed as fully differentiated and without regenerative potential. Evidence currently supports the existence of limited regenerative potential of the mature heart. Considerable current effort is being devoted to evaluating the utility of various putative stem cell populations and regenerative approaches to enhance cardiac repair after injury. The success of such approaches would offer the exciting possibility of reconstructing an infarcted or failing ventricle (Chaps. 88 and 90e).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


