Enterohemorrhagic and other Shigatoxin-producing Escherichia coli
Sivapriya Kailasan Vanaja1, Dakshina M. Jandhyala2, Emily M. Mallick3, John M. Leong1 and Sowmya Balasubramanian1, 1Tufts University School of Medicine, Boston, MA, USA, 2Division of Geographic Medicine and Infectious Disease Tufts-New England Medical Center, Boston, MA, USA, 3University of Massachusetts Medical School, Worcester, MA, USA
Background
Definition and classification
Shiga toxin-producing Escherichia coli, known as STEC, produce a typical AB5 toxin, with a single A (enzymatically active) subunit and five B (binding) subunits and characterized by its profound and irreversible cytopathic effect on Vero cells (Paton and Paton, 1998). Enterohemorrhagic E. coli, known as EHEC, is a subset of STEC that harbors additional virulence factors, such as a type III secretion system (T3SS) that translocates bacterial effectors into mammalian cells, and a large virulence plasmid (Levine, 1987). In addition to the characteristic virulence factor armament, EHEC are also defined by their ability to induce characteristic attaching and effacing (AE) lesions in the host cells (see below) and to cause manifestations such as hemorrhagic colitis and hemolytic uremic syndrome (HUS) in humans (Croxen and Finlay, 2010). Similar to the related pathogen enteropathogenic E. coli (EPEC; Chapter 4), the T3SS is encoded on the LEE (locus of enterocyte effacement) pathogenicity island, and is responsible for the AE lesions induced by EHEC (Elliott et al., 1998; Croxen and Finlay, 2010). E. coli O157:H7 is the predominant serotype of EHEC that causes human infections globally (Tarr et al., 2005; Pennington, 2010). The lack of β-glucuronidase activity (GUD–) and the inability to ferment sorbitol (SOR–) differentiate STEC O157:H7 from other E. coli strains (Paton and Paton, 1998).
STEC are characterized by enormous strain diversity. They are classified into four groups, EHEC1, EHEC2, STEC1, and STEC2 (Whittam, 1998), based on multi-locus enzyme electrophoresis. STEC O157:H7 and its evolutionary ancestor, O55:H7, constitute EHEC1. EHEC2 is the most common group of non-O157 Stx-producing strains and is comprised of serotypes such as O111:H8, O111:H-, O26:H11, and O111:H11. STEC1 is highly diverse; the common serotypes in this group are O113:H21, OX3:H21, and O91:H21. Compared with the other groups, little is known about the virulence of STEC2, which is composed of serotypes O103:H2, O103:H6, and O45:H2 (Whittam, 1998). Serotype O157:H7 and six ‘non-E. coli O157’ serogroups O26, O111, O103, O121, O45, and O145 are highly significant contributors to human disease in the US (Brooks et al., 2005).
History
In 1977, almost 100 years after Stx was originally identified in culture extracts of Shigella dysenteriae, two groups independently demonstrated that certain E. coli strains produce cytotoxins that can kill Vero cells and are neutralized by anti-Stx serum (Konowalchuk et al., 1977; O’Brien and LaVeck, 1983). These cytotoxins were termed Shiga-like toxins, owing to their striking similarity to Stx of Shigella. In 1982, following two hemorrhagic colitis outbreaks in Oregon and Michigan in the US (Centers for Disease Control, 1982), STEC came to the attention of scientists and the public alike as a prominent human pathogen. During these food-borne disease outbreaks, serotype E. coli O157:H7 was isolated from both human patients and from frozen ground beef patties. Shortly afterwards, in a seminal study, Karmali and colleagues reported the presence of cytotoxins and cytotoxin-producing E. coli in the stools of children affected with HUS (Karmali et al., 1983). Subsequent studies led to the identification of Stx and E. coli O157:H7 as the causative agents of post-diarrheal HUS. The following years were marked by several outbreaks of STEC, predominantly E. coli O157:H7, in the US, Europe, and Japan.
Evolution
STEC evolution is characterized by the acquisition of numerous virulence factors through horizontal gene transfer effected predominantly by bacteriophage transduction, allowing further rapid gene gain or loss through duplication or deletion (Kaper et al., 2004). E. coli O157:H7 is believed to have diverged from a common ancestor with E. coli K-12 about 4.5 million years ago (Reid et al., 2000). Interestingly, an atypical EPEC serotype, O55:H7, is considered the most recent precursor of E. coli O157:H7 (Whittam, 1998; Reid et al., 2000).
The emergence of E. coli O157:H7 has been analyzed extensively by several groups, leading to the formulation of a step-wise evolutionary model for this pathogen. E. coli O157:H7 strains are highly clonal in nature and are distinguished from other E. coli strains by their SOR– and GUD– phenotype. The O55:H7 SOR+ GUD+ strains evolved from ancestral EPEC-like strains through the acquisition of the LEE pathogenicity island (Figure 5.1). This was followed by the acquisition of Stx2 phage through transduction of a toxin-converting bacteriophage. The next step was marked by the gain of the EHEC virulence plasmid and an antigenic shift from O55 to O157. In further steps, E. coli O157:H7 acquired Stx1 phage and lost SOR fermentation and GUD activities, resulting in the present day GUD– SOR– strain containing Stx1, Stx2, and the EHEC plasmid (Reid et al., 2000; Wick et al., 2005).
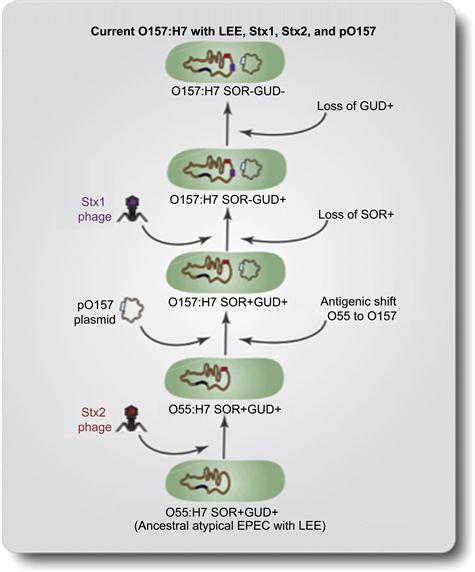
FIGURE 5.1 Step-wise model of E. coli O157:H7 evolution. In the first step, anatypical EPEC-like O55:H7 SOR+ GUD+ strain carrying the LEE pathogenicity island acquired Stx2 phage through transduction of a toxin-converting bacteriophage. In the next step EHEC virulence plasmid was gained and an antigenic shift occurred from O55 to O157. Subsequently, O157:H7 acquired Stx1 phage and lost the SOR fermentation and GUD activities, resulting in the current O157:H7 GUD– SOR– strain containing Stx1, Stx2, and the EHEC plasmid (Modified from Kaper et al., 2004).
E. coli O157:H7 is believed to be continuously evolving by mutating existing genes and by acquiring novel virulence factors, mostly by means of gene transfer through bacteriophage transduction. Several T3SS effectors in EHEC are phage-encoded, and recent studies have found that phage-encoded regulators also influence the expression of virulence factors such as T3SS (Tree et al., 2011; Flockhart et al., 2012). Advances in sequencing and molecular typing methods, such as multi-locus sequence typing, have made feasible the documentation of the ongoing evolution of this organism. Using whole genome sequencing and single nucleotide polymorphism (SNP) analysis of clinical isolates of STEC O157, several recent studies suggest the emergence of a highly virulent clade of STEC O157 that has gained new virulence genes (Manning et al., 2008). Strains belonging to this new clade (clade 8) were responsible for recent outbreaks in the US associated with leafy vegetables, all of which led to high rates of hospitalization and HUS. Clade 8 strains were also implicated in a recent outbreak in Japan (Yokoyama et al., 2011).
Thus, the ability of E. coli strains to acquire virulence factors through horizontal gene transfer continues to generate novel and unusual STEC pathotypes with increased pathogenicity. A clear example is the highly virulent Shiga toxin-producing enteroaggregative E. coli O104:H4, the causative agent of the recent German STEC outbreak (see Chapter 11). This strain is believed to have emerged as a result of transduction of enteroaggregative E. coli (EAEC) with Stx phage (Rasko et al., 2011). This hybrid strain therefore apparently has the capacity to effectively colonize the human gastrointestinal tract and the ability to produce Stx, making it exceptionally virulent. It is clear that the continuous evolution of STEC can produce new public health threats. A thorough understanding of the extent to which the genomic plasticity of STEC contributes to its pathogenicity and evolution is necessary for our ability to rapidly respond to emerging and potentially deadly STEC strains. For more details on E. coli evolution, see Chapter 3.
Epidemiology and global impact
STEC infection is a notifiable disease in the US and several other countries (Chang et al., 2009). It is highly prevalent in the US, countries of the European continent, the UK, Japan, Argentina, and Australia, although the predominant serotype varies with location (Table 5.1). In the US, following the first outbreak in 1982, the number and severity of STEC outbreaks has dramatically increased. According to the CDC (Centers for Disease Control and Prevention, 2012), STEC causes approximately 265,000 cases of illness, including 3,600 hospitalizations, and 30 deaths in the US each year (Scallan et al., 2011). E. coli O157:H7 is the predominant STEC serotype responsible for human disease in the US (Tarr et al., 2005), causing more than 96,000 cases of illness and 17 outbreaks annually. Between 1982 and 2002 there were 350 reported outbreaks of E. coli O157:H7 in the US. Of those affected, 17% were hospitalized, 4% developed HUS, and 0.5% suffered lethal infection (Scallan et al., 2011). In 2011 alone, STEC O157 caused three of the four multistate STEC outbreaks in the US. While the transmission route has been primarily (52%) foodborne, other routes of transmission such as waterborne, person-to-person, and animal contact have also caused outbreaks. Ground beef has been the major food transmission vehicle (causing 41% of outbreaks), followed by produce (21%) (Rangel et al., 2005). However, while adoption of stringent control measures has resulted in a decrease in ground beef-associated outbreaks in the US (Rangel et al., 2005; CDC, 2007) fresh produce, particularly green leafy vegetables such as spinach and lettuce, have been increasingly linked to STEC outbreaks in recent years (see below).
TABLE 5.1
STEC serogroup distribution across the world
| Country/Continent | Prevalent serogroups | Predominant serogroup O157 vs non-O157 |
| United States | O157, O26, O111, O103, O121, O45, O145 | O157 |
| Canada | O157, O55, O125, O26, O126, O128, O18 | O157 |
| South America | O1, O2, O15, O25, O26, O49, O92, O11 | Non-O157 |
| United Kingdom | O157 | O157 |
| Continental Europe | O157, O26, O111, O104, O103, O128, O91, O113, O2, O9, O145 | Both |
| Australia | O157, O111, O26 | Both |
| Japan | O157, O26, O111 | O157 |
Although the E. coli O157:H7 serotype remains most commonly associated with STEC disease in the US, a few recent outbreaks have brought non-E. coli O157 serotypes of STEC into focus (Brooks et al., 2005; Bosilevac and Koohmaraie, 2011; Bradley et al., 2012). Indeed, the burden of illness resulting from non-E. coli O157 serotypes may not be fully recognized because of diagnostic limitations and inadequate surveillance. Non-E. coli O157 strains have been estimated to cause approximately 160,000 cases of illness annually in the US (Scallan et al., 2011). Major non-E. coli O157 STEC serotypes isolated in the US between 1983–2002 include O26 (22%), O111 (16%), O103 (12%), O121 (8%), O45 (7%), and O145 (5%) (Brooks et al., 2005).
In Canada, as in the US, E. coli O157:H7 is the predominant serotype, causing 93% of STEC infections (Pennington, 2010). Other major serotypes isolated from humans in Canada include O55, O125, O26, O126, O128, and O18 (Table 5.1). The incidence of STEC in Canada ranges from 3.0 to 5.3 cases per 100 000, peaking in 1989 and declining thereafter (CCDR, 1997). Ground beef has been the most common source of outbreaks, followed by raw milk, apple juice, and contaminated water.
In contrast to the North American countries, STEC epidemiology in South America is marked by a strikingly low incidence of the E. coli O157:H7 serotype (Lopez et al., 1995, 1998; Aidar-Ugrinovich et al., 2007). For example, E. coli O157:H7 was isolated from 0–18% of STEC cases in Argentina, 9% in Chile, and none in Uruguay. Common serotypes isolated from HUS and bloody diarrhea cases include E. coli with serotypes O1:NM, O2:NM, O15:H(–), O25:H(–), O26:H(–), O49:H10, O92:H3, and O11:NM (Table 5.1). South America, specifically Argentina, has an exceptionally high HUS frequency (22 cases per 100,000 children less than 5 years old) (Lopez et al., 1998).
STEC infections are a very important public health problem in the UK, causing 1% of all food poisoning cases in England and Wales and 3% in Scotland (Money et al., 2012). In the UK, O157:H7 is the most common STEC serotype; other serotypes of STEC are rarely isolated from human cases. E. coli O157:H7 was isolated first in the UK from an outbreak of HUS in 1983 (Taylor et al., 1986). In contrast to the UK, both STEC O157 and non-E.coli O157 serotypes are equally important in Continental Europe, especially in Germany where STEC O157 causes only 30.5% of STEC infections (Eurosurveillance Editorial, 2012). However, compared to the US and the UK, STEC outbreaks are observed less frequently in Continental Europe. In 2010, a total of 4000 cases were reported in the European Union (EU), with Germany accounting for 56.8% and the Netherlands accounting for 22.0% (Eurosurveillance Editorial, 2012). Common non-E. coli O157 serotypes isolated from the EU include serotypes O26, O111, O103, O128, O91, O113, O2, O9, and O145 (Pennington, 2010) (Table 5.1). It is important to note that in the EU in 2011, an unusual serotype of STEC, O104:H4, caused one of the deadliest known STEC outbreaks ever recorded (∼3.3% mortality rate). This outbreak originated in Germany in May 2011 and later spread to most of the countries in Europe. Approximately 4200 cases were identified globally. This outbreak was marked by a strikingly high rate of HUS (22%) and hemorrhagic colitis (78%) (Buchholz et al., 2011; Rasko et al., 2011; Wu et al., 2011).
The overall yearly rate of STEC infections in Australia is markedly lower than in North American and European countries: only 0.4 cases per 100,000 between 2000–2010, of which 0.12 was STEC O157 serotype (Vally et al., 2012). Thus, STEC O157 and non-E.coli O157 serotypes have been isolated at similar rates from Australian STEC patients (Table 5.1). The predominant non-E.coli O157 serotypes are O111 (13.7%) and O26 (11.1%) (Vally et al., 2012). New Zealand, Australia’s nearest neighbor has a higher rate, e.g. 3.3 cases per 100,000 in 2009 (Leotta et al., 2008; Vally et al., 2012).
Among Asian countries, STEC disease is most prevalent in Japan (0.74 cases per 100,000 in 1999–2004), where multiple outbreaks occur every year (Sakuma et al., 2006). Although other serogroups, such as O26 and O111, have been reported to cause outbreaks, serogroup O157 is the predominant serogroup in Japan (Table 5.1). In 1996, multiple STEC O157 outbreaks throughout Japan affected 11 826 people and caused 12 deaths. Indeed, one of the largest reported E. coli O157:H7 outbreaks in history occurred that year in Sakai City, affecting approximately 8000 people, although with very low mortality (i.e. two deaths) (Izumiya et al., 1997). Until recently, only a few studies on STEC epidemiology have been done in other Asian countries. Human cases of STEC have been reported in India, Thailand, Vietnam, and Bangladesh. In 2006, the prevalence of STEC in Dhaka, Bangladesh was estimated to be 0.5% among hospitalized patients. Interestingly, no STEC O157 strains were isolated from these patients; the common serotypes isolated included O32:H25, O2:H45, O76:H19, ONT:H25, and ONT:H19 (Islam et al., 2007, 2008). Because of the low incidence of severe disease and complications such as HUS, the STEC strains in these developing countries are believed to be less virulent.
Economic impact
STEC outbreaks inflict a severe global economic burden both on the public and on the food industry due to costs associated with illness, product recalls, and compensations. The Economic Research Service of the USDA estimated the annual cost of illness due to STEC in the US at $1 billion in 2000. As of 2009, illness due to STEC O157 alone was estimated to cost $478 million in the US (Frenzen et al., 2005). Additionally, the total cost of each outbreak is amplified by legal costs and losses due to product recalls. For instance, a 1996 outbreak associated with an apple juice outbreak in the US resulted in $1.5 million in federal fines, $6.5 million in losses due to a product recall, and $12 million in claim settlements (Pennington, 2010). In Canada, the annual economic impact of STEC O157 illness alone is CAD$21 million, and more than CAD$82 million due to product recalls and other associated costs (George Morris Study Excerpt, 2007). Illness from STEC illnesses costs  9.1 million annually in the Netherlands (Tariq et al., 2011), and the 2011 German STEC outbreak accrued a total cost of $2.84 billion throughout the EU. Additionally, EU farmers claimed to have lost $600 million per week during the outbreak period (Kirk, 2011).
9.1 million annually in the Netherlands (Tariq et al., 2011), and the 2011 German STEC outbreak accrued a total cost of $2.84 billion throughout the EU. Additionally, EU farmers claimed to have lost $600 million per week during the outbreak period (Kirk, 2011).
Molecular pathogenesis
The molecular pathogenesis of EHEC has been the subject of intense study due not only to its global clinical significance but also its diversity of pathogenic strategies that serve as models for other bacterial infections. As outlined below, EHEC interacts with the intestinal epithelium in a highly characteristic fashion, manipulates many host cell functions by injecting dozens of bacterial effectors, regulates gene expression by sensing products of both nearby bacteria and mammalian cells, and causes life-threatening disease by the local production of a potent cytotoxin that is absorbed systemically. One potential limitation of our understanding of the pathogenesis of infection by EHEC is that the majority of the findings discussed in this chapter are derived from studies of a few E. coli O157:H7 strains. Investigation of how broadly these pathogenic features are shared by other EHEC may be a fruitful area of future investigation.
Entry
E. coli O157:H7 and other STECs are primarily transmitted to humans through consumption of contaminated foods (Bell et al., 1994; Itoh et al., 1998; Cody et al., 1999; Guh et al., 2010; Buchholz et al., 2011). The infectious dose of EHEC is extremely low (<100 organisms), indicating that it can pass through the stomach with high efficiency (Tilden et al., 1996; Nataro and Kaper, 1998; Doyle and Pariza, 2001; Yoon and Hovde, 2008). A critical aspect of successful host entry by E. coli is its ability to withstand the highly acidic (pH 1.5–3.0) gastric environment (Waterman and Small, 1996; Large et al., 2005). EHEC possess at least three acid resistance (AR) systems (AR1, AR2, and AR3) that regulate an acid resistance response (Smith et al., 1992; Stim and Bennett, 1993; Lin et al., 1995; Hersh et al., 1996; Castanie-Cornet et al., 1999) (for review see Foster (2004)). In addition, the chaperone HdeA, RNA polymerase-associated-protein SspA, small non-coding DsrA RNA, and DNA-binding protein Dps have been shown to have a role in acid resistance (Lease et al., 2004; Hansen et al., 2005; Kern et al., 2007; Calhoun and Kwon, 2011; Hong et al., 2012).
Adherence
Initial attachment
Upon transit through the stomach, EHEC must adhere to the luminal surface of the large intestine in order to effectively colonize the host and compete with normal microbiota. EHEC encodes a multitude of surface factors including fimbrial and non-fimbrial adhesins (Table 5.2; for review, see Torres et al. (2005) and Bardiau et al. (2010)) that may mediate the initial interaction with the host mucosa and promote colonization and disease (La Ragione et al., 2000; Perna et al., 2001; Cergole-Novella et al., 2007).
TABLE 5.2
| Potential adhesins | Function |
| EspA | Filamentous appendage that promotes type III effector delivery and mediates interaction between host cells and AE pathogens (Ebel et al., 1998; Knutton et al., 1998; Daniell et al., 2001). |
| Long polar fimbriae (LPF) | Required for adherence and microcolony formation in eukaryotic cells (Torres et al., 2002; Torres et al., 2004). Has a role in pig, sheep (Jordan et al., 2004) and infant rabbit colonization (Lloyd et al., 2012) and influencing intestinal tissue tropism (Fitzhenry et al., 2006). Binds to ECM proteins (Farfan et al., 2011). |
| F9 Fimbriae (Type I pilus homolog) | Potential role in colonization in young calves and in adherence to cells in culture (Low et al., 2006). |
| Curli fimbriae | Associated with increased virulence of EHEC in mice (Uhlich et al., 2002). Promotes attachment of EHEC (O111:H-7-57C+, O157:H7 5-11C+ and O103:H2 7-52C+) to certain abiotic surfaces (Cookson et al., 2002; Pawar et al., 2005). |
| OmpA | Promotes adherence to HeLa and polarized Caco-2 cells (Torres and Kaper, 2003; Torres et al., 2006). |
| IrgA homolog adhesion (Iha) | Confers the adherence phenotype on laboratory E. coli; possess siderophore-receptor activity (Tarr et al., 2000; Rashid et al., 2006). |
| EHEC factor for adherence (Efa) | Important for adhesion of non-O157 EHEC to CHO cells in vitro, hemagglutination, autoaggregation and colonization of bovine intestines (Nicholls et al., 2000; Stevens et al., 2002; Deacon et al., 2010). |
| Calcium binding Antigen 43 homolog (Cah) | Binds to calcium and promotes autoaggregation and biofilm formation (Torres et al., 2002). |
| Hemorrhagic coli pilus (HCP) (Type IV pili) | Binds to laminin and fibronectin, promotes twitching motility, biofilm formation and hemagglutination (Xicohtencatl-Cortes et al., 2007, 2009). Activates release of pro- and anti-inflammatory cytokines from polarized, cultured intestinal cells (Ledesma et al., 2010). |
| Escherichia coli common pilus (ECP) | Important for adherence to cultured epithelial cells and virulence of E. coli O157 in humans (Rendon et al., 2007). |
| ToxB | Homolog of Efa1, encoded on plasmid pO157 and promotes adherence to cultured epithelial cells (Tatsuno et al., 2001). Affects production and secretion of some virulence factors required for the development of A/E lesions (Stevens et al., 2004). |
| Porcine attaching-effacing associated protein (Paa) | Required for the A/E activity by many A/E strains, including O157:H7 and promotes initial bacterial adherence (An et al., 1999; Batisson et al., 2003). |
| STEC autoagglutinatingadhesin (Saa) | Plasmid encoded and isolated from LEE-negative STEC (Paton et al., 2001). Saa-harboring STEC exhibit differential binding properties to epithelial cells (Toma et al., 2008). Saa positive STEC frequently isolated from bovine rather than human origin (Jenkins et al., 2003). |
| Flagella | O157:H7 flagella agglutinates rabbit red blood cells, promotes adherence to mucins I and II and persistence in chicken in the absence of intimin (Best et al., 2005; Erdem et al., 2007). |
| AdfO | Secreted from T2SS and encoded on the cryptic prophage CP-933O, AdfO promotes for adherence of EHEC to cultured HeLa cells (Ho et al., 2008). |
| YodA | AdfO facilitates YodA secretion from pO157-encoded T2SS and promotes adherence of EHEC to HeLa cells and colonization of the infant rabbit intestine (Ho et al., 2008). |
| StcE | A zinc metalloprotease secreted by the T2SS on the plasmid pO157, cleaves serpin C1 esterase inhibitor (Lathem et al., 2002, 2004). Reduces the mucous glycocalyx cell surface barrier and promotes a closer interaction between the bacterium and host cell (Grys et al., 2005, 2006). |
The EHEC-specific long polar fimbriae (LPF) (Torres et al., 2002a, 2004) was recently shown to be involved in intestinal colonization of infant rabbits (Lloyd et al., 2012). Additionally, bacteria deficient in production of hemorrhagic coli pilus (HCP), a type IV pilus that EHEC shares with most other E. coli (see Chapter 13), display a significant reduction in adherence to bovine gut explants (Xicohtencatl-Cortes et al., 2007). Moreover, HCP generates an antibody response in patients with HUS, suggesting that these pili are produced during infection (Xicohtencatl-Cortes et al., 2007). EspA, which forms a filamentous appendage from EHEC’s T3SS, has been suggested to be an adhesin promoting initial interaction between host cells and bacteria (Ebel et al., 1998). YodA and StcE, two effectors secreted through a type II secretion system, promote EHEC adherence to cultured monolayers, as well as during colonization of infant rabbits (Grys et al., 2005; Ho et al., 2008). Non-fimbrial EHEC adhesins include OmpA, and have been shown to facilitate bacterial binding to cultured monolayers (Torres and Kaper, 2003). Finally, H7 flagella have been hypothesized to promote adherence to mucus and extracellular matrix proteins within the host intestinal tract (Erdem et al., 2007).
The best-characterized adhesin of EHEC, and an absolutely essential virulence factor, is the ∼94-kDa outer-membrane protein intimin, encoded by the eae gene (Jerse et al., 1990; Donnenberg and Kaper, 1991; Beebakhee et al., 1992; Yu and Kaper, 1992). Intimin is an integral outer membrane protein with three domains: a flexible N-terminal region thought to be located in the periplasm, a central region that forms a β-barrel in the outer membrane, and a surface-exposed C-terminal receptor-binding domain (Ross and Miller, 2007; Yi et al., 2010). The latter domain exhibits considerable allelic variation (Frankel et al., 1994), with at least ten different intimin subtypes (Adu-Bobie et al., 1998; Oswald et al., 2000).
Intimin was first identified by its essential role in the formation of the characteristic attaching and effacing (AE) lesions on the surface of intestinal epithelial cells (Jerse et al., 1990; Donnenberg and Kaper, 1991). AE lesions are characterized by effacement of microvilli, intimate bacterial attachment to epithelial cells, and the formation of localized actin assembly, i.e. filamentous (F–) actin ‘pedestals’, beneath bound bacteria (Staley et al., 1969; Ulshen and Rollo, 1980; Moon et al., 1983). Intimin promotes AE lesion formation by binding to Tir (translocated intimin receptor), a type III-secreted effector that localizes in the host plasma membrane after translocation into mammalian cells (see below) (Kenny et al., 1997b; Hartland et al., 1999). Thus, intimin is required for tight attachment to cultured cells, as well as for colonization and virulence during mammalian infection (Donnenberg et al., 1993b; McKee et al., 1995; Tzipori et al., 1995; Dean-Nystrom et al., 1998).
Intimin may possess Tir-independent functions as well. Intimin of the related EPEC has been shown to contribute to the disruption of epithelial barrier function of polarized monolayers in a Tir-independent manner (Dean and Kenny, 2004), a function that may be related to Tir-independent host cell attachment. Indeed, intimin promotes binding to cultured cells in the absence of Tir, albeit with much lower efficiency than in the presence of Tir binding. (Frankel et al., 1994; McKee and O’Brien, 1995; Frankel et al., 1998a, b; Hartland et al., 1999). Several host cell receptors, including β1-chain integrins (Frankel et al., 1996; Sinclair et al., 2006) and nucleolin (Sinclair and O’Brien, 2002; Sinclair et al., 2006) have been shown to have the capacity to bind intimin. Interestingly, Stx2 (see below) has been shown to enhance adherence of EHEC to intestinal epithelial cells and intestinal colonization in mice by increasing the surface localization of nucleolin (Robinson et al., 2006; Liu et al., 2010). Whereas all alleles of intimin bind to Tir, alleles differ in promoting epithelial colonization in some experimental models, consistent with a Tir-independent colonization function (Tzipori et al., 1995; Phillips and Frankel, 2000; Fitzhenry et al., 2002; Girard et al., 2005; Mundy et al., 2007). Finally, intimin of the murine pathogen Citrobacter rodentium, which also generates AE lesions (see below), plays a Tir-independent role in colonization of streptomycin-pretreated mice (Mallick et al., 2012a).
Formation of attaching and effacing lesions
The best-characterized interactions between EHEC and gut epithelia involve the formation of AE lesions, described above, which are characteristic of EHEC and the other AE pathogens EPEC and C. rodentium. AE lesion formation requires Tir, which is translocated to the host cell by a T3SS, a multi-protein export apparatus that spans the inner and outer membranes and facilitates direct injection of bacterial effectors from the bacterial cytoplasm into the host cell (see Chapter 14; Garmendia et al., 2005). Tir, intimin, and the entire T3SS are encoded on a ∼35 kb chromosomal pathogenicity island called the Locus of Enterocyte Effacement (LEE) (McDaniel et al., 1995).
Tir, like intimin, is absolutely required for AE lesion formation and colonization in many animal models (Marches et al., 2000; Deng et al., 2003; Ritchie et al., 2003). Once injected into the host cell, Tir integrates into the host cell membrane in a hairpin loop conformation, with its central extracellular domain serving as the receptor for the C-terminal extracellular domain of intimin (Batchelor et al., 2000; Luo et al., 2000; Liu et al., 2002). Intimin-binding by Tir induces Tir clustering (Touze et al., 2004) and permits the cytoplasmic domains of Tir to coordinate a downstream signaling cascade resulting in the formation of F-actin pedestals (Campellone et al., 2006; Hayward et al., 2006; Brady et al., 2007).
A cytoplasmic activity of Tir critical for F-actin pedestal formation is recognition by mammalian adaptor proteins IRTKS (Insulin Receptor Tyrosine Kinase Substrate) and/or IRSp53 (Insulin Receptor Substrate Protein 53 kDa) (Vingadassalom et al., 2009; Weiss et al., 2009; de Groot et al., 2011). These homologous proteins encode an I-BAR (membrane-binding) and an SH3 (polyproline-binding) domain, and can stimulate low-level actin assembly. However, in EHEC strain O157:H7, robust actin pedestal formation requires a second-type III-secreted effector, EspFU/TccP, which is encoded on the cryptic prophage CP-933U/Sp14 (Campellone et al., 2004; Garmendia et al., 2004), and which is recruited to sites of bacterial binding by the IRTKS/IRSp53 SH3 domain. In turn, EspFU/TccP binds directly to the GTPase binding (GBD) region of N-WASP, resulting in its activation (Cheng et al., 2008; Sallee et al., 2008; Campellone, 2010) and the subsequent activation of the actin nucleator Arp 2/3 (reviewed in Goley and Welch (2006) and Stradal and Scita (2006)), finally resulting in actin pedestal formation.
Mutants of EHEC or other AE pathogens that entirely lack intimin or Tir are incapable of generating AE lesions and unable to colonize a range of mammalian hosts (Donnenberg et al., 1993a; Tzipori et al., 1995; Marches et al., 2000; Deng et al., 2003; Ritchie et al., 2003). Given that intimin- or Tir-deficient mutants are almost entirely defective for mammalian cell attachment, their inability to colonize animals may reflect a binding defect rather than a pedestal formation defect per se. Instead, the potential role of actin assembly in the pathogenesis of disease requires the analysis of mutants of AE pathogens that express intimin and Tir and are competent for attachment but are specifically defective for actin assembly. In fact, a mutant with this phenotype, i.e. an EHEC espFU/tccP mutant, is defective for colonization of infant rabbits at late time points and forms unusually small bacterial aggregates in intestines of gnotobiotic piglets (Ritchie et al., 2008). Additionally, C. rodentium mutants similarly specifically defective in generating actin pedestals have diminished colonization ability during coinfection with wild-type bacteria (Crepin et al., 2010), or were found to be deficient in colonizing the colonic mucosa and causing lethal disease in an Stx-producing C. rodentium model (E. Mallick, unpublished observation). These findings indicate that pedestal formation by EHEC promotes colonization and stx-mediated disease.
Regulation of gene expression
Expression of major EHEC virulence factor genes, such as stx, the large pO157 plasmid, and the LEE, is regulated by multiple genes and environmental signals. (A detailed description of the highly complex pathways of EHEC regulation is beyond the scope of this chapter and readers are referred to recent reviews (Mellies et al., 2007; Croxen and Finlay, 2010)). Regulation of the LEE, which is comprised of 41 open reading frames arranged in five major polycistronic operons, LEE1 through LEE5, has been the subject of considerable investigation. An important element of LEE regulation involves the global nucleoprotein-like regulator H-NS, which acts as a major suppressor of LEE expression (Bustamante et al., 2001). Ler, the major LEE-encoded regulator, is central to the control of LEE expression, and disrupts H-NS-mediated silencing of LEE (Mellies et al., 2007), leading to increased transcription of operons LEE2 through LEE5. The additional LEE-encoded regulators include GrlA (global regulator of LEE activator) and GrlR (global regulator of LEE repressor). GrlA binds to Ler promoter and acts as an H-NS antagonist thereby promoting the expression of Ler (Jimenez et al., 2010). GrlR interacts with GrlA, interfering with the GrlA-mediated induction of Ler leading to an inhibition of LEE expression (Iyoda et al., 2006).
LEE expression is also controlled by regulators encoded elsewhere on the EHEC genome. Some of these, such as Pch (Iyoda and Watanabe, 2004), IHF (Yona-Nadler et al., 2003), RcsCDB and GrvA (Tobe et al., 2005) induce LEE expression by increasing Ler production. Negative regulators of LEE, such as GadE, GadF (Tatsuno et al., 2003; Kailasan Vanaja et al., 2009), EtrA and EivF (two regulatory proteins of E. coli type III secretion system 2, ETT2) (Zhang et al., 2004), Hha (Sharma and Zuerner, 2004), CadA (Vazquez-Juarez et al., 2008), and SdiA (Kanamaru et al., 2000; Sharma et al., 2010) regulate LEE expression mainly by inhibiting Ler. Finally, the sRNA chaperone Hfq regulates the LEE in a strain-specific manner, indicating that the LEE is subject to complex regulation by sRNA (Hansen and Kaper, 2009).
During intestinal infection, EHEC is exposed to extracellular factors, such as EHEC-secreted components, products of the existing microbiota, and host-derived signals. Several extracellular factors that regulate LEE expression by targeting some of the above pathways have been investigated. Growth of EPEC and EHEC in Dulbecco’s modified Eagle’s medium (DMEM) at host body temperature (37°C), pH 7.0, and physiological osmolarity induces LEE expression, an observation that has been historically utilized to facilitate investigation of the T3SS (Kenny and Finlay, 1995; Kenny et al., 1997a). Interestingly, expression of EHEC LEE is regulated by a quorum-sensing system that recognizes both the bacterial autoinducer AI-3 and host-derived catecholamines. This inter-kingdom communication involves multiple two-component systems that positively and negatively regulate both the LEE and non-LEE-encoded type III effectors (Kendall and Sperandio, 2007; Reading et al., 2007; Hughes and Sperandio, 2008). Other extracellular factors critical to LEE expression include iron, sodium bicarbonate, ammonium chloride, calcium, glucose, and short-chain fatty acids, which are end-products of dietary carbohydrate fermentation whose concentrations in the intestine vary depending on the microbiota and nutrient conditions (Abe et al., 1997; Ide et al., 2003; Herold et al., 2009; Delcenserie et al., 2012).
Although clearly the LEE encodes a central virulence attribute of EHEC O157:H7, many other loci contribute to the virulence of this bacterium. Several of the regulatory pathways that control LEE expression also function as regulators of other virulence factors. For example, Ler increases expression of long polar fimbriae and the secreted protease StcE encoded on the pO157 virulence plasmid (Lathem et al., 2002; Torres et al., 2007). The quorum-sensing system that recognizes the bacterial autoinducer AI-3 and host catecholamines also regulates Shiga toxin and flagellae production in a complex manner (Sperandio et al., 2002; Jeon and Itoh, 2007).
Disruption of host defense
Immune responses may promote clearance of the bacterium, or could facilitate host damage, and an important component of the pathogenesis of EHEC and other AE pathogens is the ability to disrupt bacterial recognition by the innate immune system. It has been suggested that EHEC/EPEC infection blocks phagocytosis, one of the first steps in the development of an antimicrobial immune response. T3SS proteins such as EspB, EspF, EspH, and EspJ are implicated in this antiphagocytic activity of EHEC/EPEC (Wong et al., 2011). EspB binds to the actin binding domains of several myosin family members and may block the closure of the phagocytic cup (Diakonova et al., 2002; Iizumi et al., 2007). EspF also appears to inhibit phagocytosis by binding to actin (Alto et al., 2007). Additionally, EspH blocks phagocytosis by binding and inhibiting DH-PH domain-containing Rho GTPases, which control cytoskeletal remodeling during phagocytosis (Dong et al., 2010). In contrast, the mechanism of EspJ-mediated antiphagocytic activity remains unknown even though it has been shown to inhibit two phagocytic pathways mediated by FCγR and CR3 (Marches et al., 2008).
EHEC/EPEC further disrupts mounting of an effective proinflammatory response by interfering with innate immune signaling pathways. A common target in the host for this purpose is the nuclear factor kappa B (NF-κB), a key proteinin expression, and production of proinflammatory cytokines. Normally, the NF-κB-inhibitory protein IκB sequesters NF-κB in an inactive state, and upon activation of pattern recognition receptors such as TLRs by pathogen-associated molecular patterns (PAMPs), active NF-κB is released from IκB and translocates from the cytosol to the nucleus to activate transcription of target genes, such as those encoding proinflammatory cytokines and chemokines (Kawai and Akira, 2007). These proinflammatory mediators potentially promote clearance of the infection.
Some of the most detailed studies addressing disruption of inflammatory signaling in the host cell involve the action of non-LEE encoded (Nle) type-III-secreted effectors such as NleE, NleB, NleC, NleD, and NleH. Partly because type III translocation by in vitro cultivated EPEC is more efficient than that by EHEC, work on type III effectors shared by the two pathogens has been performed largely in EPEC. Given the high sequence homology of EPEC and EHEC (and C. rodentium) effector alleles, it seems likely that the mechanisms of action are conserved among all the AE pathogens (see also Table 5.3). NleE appears to inhibit TNFα-induced NF-κB activation by inhibiting its nuclear translocation (Zurawski et al., 2008; Newton et al., 2010; Vossenkamper et al., 2010). Recent studies indicate that NleE modifies host ubiquitin chain sensory proteins, TAB2/3, to disrupt host NF-κB signaling (Zhang et al., 2012). NleE also prevents degradation of IκB by inhibiting activation of IKKbeta resulting in further NF-κB suppression (Nadler et al., 2010). NleB, has been shown to enhance this particular activity of NleE (Nadler et al., 2010). Furthermore, NleC and NleD have been shown to cleave the p65 subunit of NF-κB and c-Jun N-terminal kinase (JNK), preventing inflammatory responses (Yen et al., 2010). Additionally, EHEC homologs NleH1 and NleH2 also can bind to NF-κB subunit RPS3 (Wan et al., 2011). All these findings demonstrate that EHEC/EPEC has evolved a strategy to interfere with the induction of a proinflammatory response by simultaneously blocking multiple steps in the inflammatory signaling cascade through the concerted activity of several T3SS effector proteins.
Host damage
During infection, AE pathogens attach to the luminal surfaces of the host intestinal epithelia where they efface localized regions of microvilli, form actin pedestals, and translocate effector proteins into the host cell. Many of these effector proteins have been shown to subvert various cellular processes and dramatically alter the function of intestinal cells, influencing colonization and likely contributing significantly to the development of diarrhea (for review see Frankel et al. (1998a) and Wong et al. (2011)).
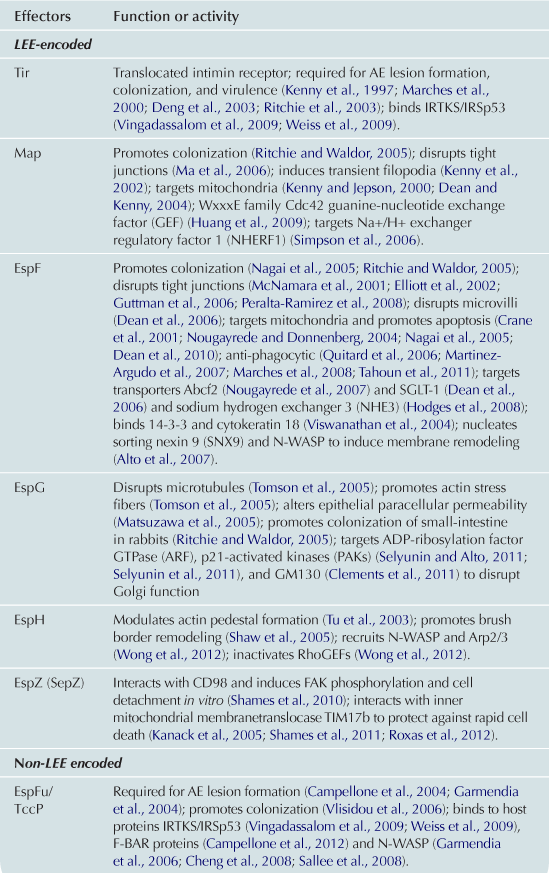
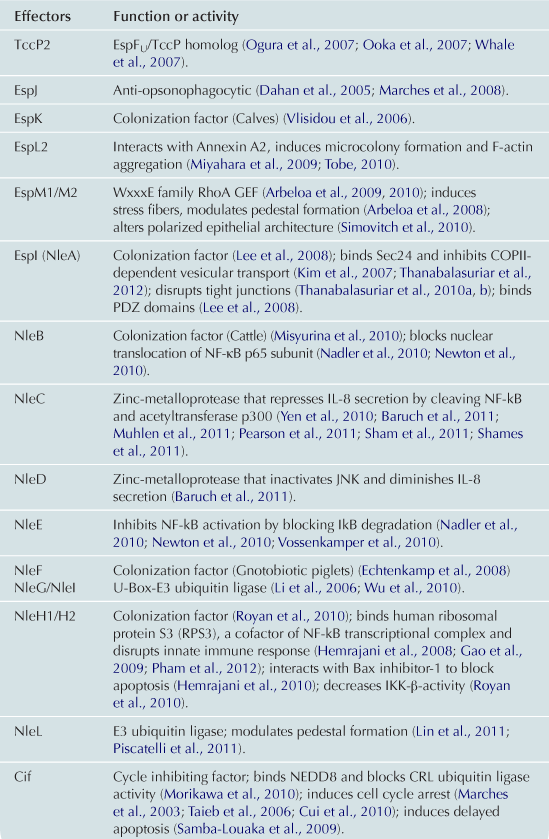
Translocated effectors are responsible for much of the damage to host cells. The precise number of effectors has not been definitively determined, but certainly numbers in the dozens (Tobe et al., 2006). The set of effectors includes those that (i) disrupt actin cytoskeleton, resulting in the formation of pedestals (see above), the disruption of tight junctions and/or microvilli; (ii) interfere with vesicle trafficking pathways; (iii) induce mitochondrial dysfunction; or (iv) trigger apoptosis. Given that many effectors are described in some detail in Chapters 4 and 15 of this volume, in this narrative we give a relatively brief overview of some of the critical effectors. In addition, Table 5.3 describes the in vitro and in vivo functions of many of the EHEC effector proteins, as well as references to primary literature.
Alteration of host vesicle trafficking pathways
EHEC and other AE pathogens manipulate host vesicle trafficking pathways. The effector EspG binds several eukaryotic proteins, including ADP-ribosylating GTPase (ARF), which regulates Golgi membrane trafficking, the Golgi matrix protein GM130, and p21-activated kinases (PAKs), which in turn regulate a wide variety of cellular processes (Clements et al., 2011; Selyunin et al., 2011b). Consistent with the hypothesis that EspG functions as a scaffold to generate a novel signaling complex on the Golgi membrane, EspG localizes to Golgi, inhibits endomembrane trafficking, and induces Golgi fragmentation. NleA inhibits vesicle trafficking between the endoplasmic reticulum and the Golgi and interacts with the Sec24 subunit of the COPII vesicle complex, which controls membrane fusion events (Kim et al., 2007; Lee et al., 2008a; Thanabalasuriar et al., 2012). Finally, overexpression of EspF (E. coli secreted protein F) in mammalian cells induces membrane tabulation (Alto et al., 2007) and like EspG, appears to function as a scaffold to generate a novel complex by targeting more than one eukaryotic target, in this case SNX9, which binds to membranes and regulates endocytosis, and the actin nucleation promoting factor N-WASP (Weflen et al., 2010).
Disruption of epithelial barrier and absorptive function
Epithelial tight junctions (TJs) serve to separate the luminal and adluminal environments as well as the apical membrane and basolateral membrane proteins. AE pathogens target tight junctions, leading to compromised barrier function that has been assayed by increased permeability and decreased transepithelial resistance of polarized monolayers (Canil et al., 1993; Dean and Kenny, 2004; Shifflett et al., 2005; Guttman et al., 2006a, b). The functional compromise of TJs correlates with morphological changes, for example the redistribution of TJ-associated proteins such as ZO-1, claudin, and occludin (Simonovic et al., 2000; Roxas et al., 2010; Zhang et al., 2010). Changes in epithelial barrier function observed during infection of monolayers have also been documented during animal infection by C. rodentium (Guttman et al., 2006a; Ma et al., 2006; Flynn and Buret, 2008). The mechanism of TJ disruption is incompletely understood. The effector EspF, described above, is required for this step (McNamara et al., 2001; Dean and Kenny, 2004; Guttman et al., 2006a; Peralta-Ramirez et al., 2008), and its function can be supplied by the related effector EspFU/TccP, which plays a critical role in pedestal formation and which, like EspF, is capable of binding to the actin nucleation promoting factor N-WASP (Viswanathan et al., 2004a). Map (Mitochondrial-associated protein), which activates Rho GTPases, is also required for tight junction disruption (McNamara et al., 2001; Dean and Kenny, 2004; Viswanathan et al., 2004a; Simpson et al., 2006; Huang et al., 2009). Finally, intimin (but not Tir) plays a role in this activity (Dean and Kenny, 2004), although the precise function of intimin in this process is not known.
The mechanisms of effacement, i.e. the loss of absorptive microvilli of host cells, are not well understood, but intimin, at least three type III effectors, i.e. Map, EspF, and Tir, and one or more host signaling pathways dependent on the host Ca++-dependent protease calpain are required (Potter et al., 2003; Dean et al., 2006; Lai et al., 2011). As enterocyte microvilli vastly increase cell surface area and thus the absorptive capacity of the intestine, the effacement of intestinal epithelium may contribute to the diarrhea characteristic of EHEC infection.
Induction of mitochondrial dysfunction and apoptosis
In addition to their involvement in disruption of other host processes, EspF and Map also have mitochondria-associated functions. EspF localizes to mitochondria and disrupts the mitochondrial membrane potential, leading to mitochondrial swelling and damage (Kenny and Jepson, 2000; Kenny, 2002) and triggering cell apoptosis (Crane et al., 2001; Nougayrede and Donnenberg, 2004; Nagai et al., 2005). Map also is targeted to mitochondria, causing mitochondrial dysfunction both in cultured cells and in colonoctyes during murine infection by C. rodentium (Kenny and Jepson, 2000; Ma et al., 2006; Papatheodorou et al., 2006). Apoptosis can apparently be also triggered by mitochondrial-independent means, as another effector, Cif, impairs the ubiquitin proteosome system, leading to disruption of the cell-cycle (Marches et al., 2003) and delayed apoptosis (Samba-Louaka et al., 2009; Jubelin et al., 2010).
Shiga toxins and development of HUS
Infection with Shiga toxin-producing Escherichia coli (STEC) can result in a microangiopathic sequela termed the hemolytic uremic syndrome (HUS). Defined by a triad of hemolytic anemia, thrombocytopenia, and renal failure, HUS is the most frequent cause of renal failure in children (Siegler, 2003). As mentioned above, Shiga toxin, so-named because it was first identified in Shigella dysenteriae, is the critical STEC virulence factor associated with the development of HUS during STEC infection (Paton & Paton, 1998).
Shiga toxins are classified as belonging to one of two types (or groups), Stx1 or Stx2, based on their serological neutralization profile; within the two types there are several subtypes (Scheutz et al., 2012). All Shiga toxins are proteins with an AB5 quaternary structure. The enzymatic Shiga toxin A-subunit (StxA) has N-glycosidase activity, and the five B-subunits (StxB) function to bind glycolipid cell surface receptors (reviewed in Thorpe et al. (2002)). In addition, these different toxin types share similar operon structures, in which stxA is immediately upstream of stxB. Despite these similarities, Shiga toxins of a given type or subtype often have different epidemiological profiles. For example, although data suggest that O157:H7 E. coli strains that produce Stx2 but not Stx1 are more likely to be associated with HUS and severe disease in humans (Griffin and Tauxe, 1991; Boerlin et al., 1999; Thorpe et al., 2002; Beutin et al., 2008), not all Stx2 subtypes are associated with HUS in humans.
The genes encoding Shiga toxins generally reside in the late region of either functional or cryptic lambdoid prophages (Unkmeir and Schmidt, 2000; Schmidt, 2001), and induction of the phages have been shown to result in a marked up-regulation of toxin production in both Stx1- and Stx2-expressing STEC (Wagner et al., 2001, 2002). Phage induction can be triggered by activation of the SOS response, a bacterial stress response which is activated by DNA damage (Little and Mount, 1982). Certain antibiotics have been shown to activate this response and promote transcription of stx1 and stx2 genes, production of toxin, and mortality in laboratory mice (Walterspiel et al., 1992; Kimmitt et al., 2000; Zhang et al., 2000). Furthermore, during infection, antibiotic-induced bacterial cell lysis may increase the level of free luminal Stx available for systemic absorption (Kimmitt et al., 2000; Tarr et al., 2005). Consistent with these notions, antibiotic administration has been associated with an increased risk of subsequent HUS in some studies (Bell et al., 1997; Wong et al., 2000). It has therefore been suggested that administration of antibiotics, particularly those known to activate the SOS response, should be avoided for treating STEC infection. However, recent in vitro studies suggest that certain antibiotics that do not induce Stx production in STEC may be worthy of further study (Bielaszewska et al., 2012; Corogeanu et al., 2012).
The first step in Stx-mediated intoxication is binding of the B-subunit pentamer to a glycolipid receptor on the host cell surface (Figure 5.2, Step 1). The best-studied Stx receptor is globotriaosylceramide (Gb3) (Jacewicz et al., 1986; Waddell et al., 1988; Hoffmann et al., 2010). Gb3 bound toxin is endocytosed (Step 2), and trafficked retrograde through the Golgi apparatus to the endoplasmic reticulum (ER; Step 3; Sandvig et al., 1992; Arab and Lingwood, 1998; Girod et al., 1999; Sandvig and van Deurs, 2000; Tam and Lingwood, 2007). During this process, the A-subunit is proteolytically cleaved, possibly by furin located in the endosome and/or the trans-Golgi network (Garred et al., 1995a, b; Tam and Lingwood, 2007). However, it should be noted that the A-subunit is also sensitive to cleavage by cytosolic calpain (Garred et al., 1997). The resulting A1 fragment remains linked to the A2 fragment via an intra A-subunit disulfide bond (Garred et al., 1997), and investigations into Stx1 suggest the holotoxin remains intact during translocation to the ER (Tam and Lingwood, 2007). Once in the ER, it is presumed that the disulfide bond is reduced (Step 4) based on the observation that toxins such as ricin and cholera toxin have been shown to undergo disulfide bond reduction in this compartment (Majoul et al., 1997; Tsai et al., 2001; Bellisola et al., 2004; Spooner et al., 2004). Furthermore, fluorescently labeled A- and B-subunits of Stx1 do not separate during retrograde translocation to the ER, suggesting that Stx disulfide bond reduction does not occur during transport to this compartment (Tam and Lingwood, 2007). The free A1 fragment, which possesses the N-glycosidase activity, is then translocated to the cytosol (Step 5; LaPointe et al., 2005; Yu and Haslam, 2005; Tam and Lingwood, 2007), where its N-glycosidase activity results in the cleavage of a single adenine residue (A4324) located on the α-sarcin/ricin loop of the 28S ribosomal RNA (Obrig et al., 1987; Endo et al., 1988). The depurination of the ribosome results in cessation of protein synthesis (Step 6; Endo et al., 1988; Saxena et al., 1989).
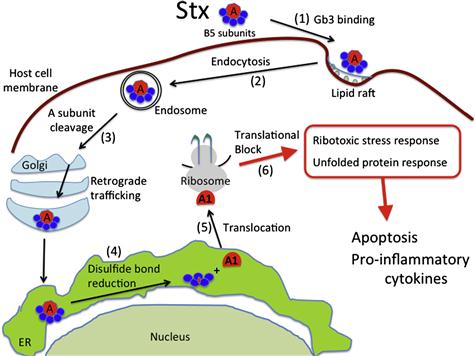
FIGURE 5.2 Generalized model of cellular intoxication by Stxs. (1) Stx holotoxin via its five B-subunits recognizes and binds to host cell Gb3 receptors localized in lipid rafts on the surface of host cells. (2) Shiga toxin undergoes endocytosis and (3) is transported retrograde through the Golgi apparatus to the lumen of the endoplasmic reticulum (ER). During this time the StxA-subunit may undergo proteolytic cleavage, possibly by furin present in the endosome or the trans Golgi network (Garred et al., 1995a,b). (4) The proteolytically cleaved StxA1 fragment is detached from the StxA2 fragment and associated StxB-subunits through reduction of an intramolecular disulfide bond. (5) The StxA1 fragment is then transported to the cytosol (Tam and Lingwood, 2007) where it depurinates the alpha sarcin loop of the 28S ribosomal RNA. (6) This damage to the ribosome results in protein synthesis inhibition, the ribotoxic stress response, and the unfolded protein response all three of which may contribute to pathology through increased inflammation and/or apoptosis.
Despite being an inhibitor of protein synthesis, in vitro studies have demonstrated that intoxication by Shiga toxin results in a paradoxical increase in cytokine expression (Sakiri et al., 1998; Thorpe et al., 1999; Foster et al., 2000; Thorpe et al., 2001). Shiga toxins have been shown to regulate cytokine expression through the ribotoxic stress response (RSR) and the phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin signaling (PI3/AKT/mTOR) pathway (Colpoys et al., 2005; Cherla et al., 2006, 2009; Jandhyala et al., 2008, 2012). Cytokines induced by Shiga toxins include interleukin-8 (IL-8), tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), and interleukin-1β (IL-1β) (van Setten et al., 1996; Thorpe et al., 1999; Cherla et al., 2006, 2009; Jandhyala et al., 2008). In addition to promoting cytokine expression, in vitro studies have also demonstrated that Shiga toxins induce apoptotic signaling via the RSR and the unfolded protein response (UPR) (Smith et al., 2003; Lee et al., 2008b).
As STEC are enteric pathogens and generally non-invasive, in order to cause systemic disease such as HUS, Shiga toxins have to pass from the intestinal lumen, where they are produced, to the systemic circulation, where the toxin can access the microvasculature of the kidney and the central nervous system. Intestinal infection of mice with a genetically engineered Stx-producing C. rodentium, which like EHEC generates AE lesions on intestinal epithelial cells, suggests that colonization of the mucosal surface may promote systemic intoxication (E. Mallick and J. Leong, unpublished observations). In vitro data have suggested that Shiga toxins may cross the epithelial barrier via a transcellular route that does not result in the death of the enterocyte (Acheson et al., 1996; Philpott et al., 1997; Malyukova et al., 2009). Alternatively, Shiga toxins may enter the systemic circulation via a paracellular route, consistent with in vitro studies suggesting that luminal infiltration of polymorphonuclear leukocytes (PMNs) may promote the basolateral uptake of Stxs (Hurley et al., 2001). Indeed, histological analyses of patients with STEC-associated hemorrhagic colitis show large infiltrates of neutrophils in the lamina propria and crypts of the large intestine (Griffin et al., 1990).
There is currently a lack of consensus as to the role Shiga toxins play in promoting intestinal damage. Murine infection with Stx-producing C. rodentium results in toxin-dependent damage to the intestinal epithelium (Mallick et al., 2012b) although it has not been determined whether Stx damages the epithelium directly or indirectly, e.g. by compromising blood supply to the mucosa. Several studies have suggested that, with the exception of the enteric endothelium and Paneth cells, the human intestine is devoid of the Stx receptor Gb3 (Holgersson et al., 1991; Berin et al., 2002; Schuller et al., 2004, 2007; Miyamoto et al., 2006). In contrast, Zumbrum et al. suggested that human colonic epithelium expresses the Stx receptor Gb3, albeit at low levels (Zumbrun et al., 2010). Whether or not Gb3 is expressed in the human colonic epithelium, Stx has been detected in epithelium from the illeocecal valves of patients infected with STEC, suggesting that Stx has the potential to enter these cells (Malyukova et al., 2009). In addition, Stx was found in expelled cells in the lumen, although whether these cells were damaged directly by Stx was not determined. It has been proposed that susceptibility to intoxication by Stx requires that Gb3 be localized to lipid rafts (Falguieres et al., 2001; Kovbasnjuk et al., 2001; Hanashima et al., 2008). It is therefore conceivable that Gb3-positive and/or -negative intestinal epithelial cells take up Stx but are not intoxicated by them.
Once in the systemic circulation, Stx can reach target organs such as the brain and kidney, where it can induce life-threatening disease. Data are inconclusive as to whether Stx circulates freely in the serum and/or is carried by blood cells to target organs. While small amounts of free Stx have been detected in HUS patients, Stx has been shown to bind in vitro to a variety of blood components including PMNs, platelets, monocytes, and red blood cells (RBCs) (Bitzan et al., 1994; van Setten et al., 1996; Te Loo et al., 2001b; Ghosh et al., 2004; Stahl et al., 2006), and Stx-positive PMNs have also been detected in patients with HUS (Brigotti et al., 2011).
HUS is a microangiopathic disorder featuring deposition of thrombi rich in fibrin and platelets in the renal microvasculature (reviewed in Blackall and Marques (2004)). Glomerular capillaries are occluded by these thrombi resulting in ischemic damage to the endothelium. Endothelial expression of IL-8, fractaline, and monocyte chemotactic protein (MCP-1) promotes endothelial adherence by leukocytes in the presence of Stx (Zoja et al., 2002; Geelen et al., 2008; Zanchi et al., 2008). Furthermore, PMNs are capable of transferring toxin to endothelial cells (Te Loo et al., 2000; Brigotti et al., 2010). Stx has been found bound to platelets (Stahl et al., 2006), PMN–platelet complexes, and monocyte–platelet complexes in HUS patients (Stahl et al., 2009). The toxin also binds to fibrinogen and promotes platelet aggregation and adherence to endothelial cells (Karpman et al., 2001), and thus could contribute to the fibrin-rich clots associated with HUS. Finally, Stx can induce the expression of tissue factor on monocytes, and trigger the release of monocyte-derived microparticles, which may contribute to thrombosis by increasing the amount of circulatory tissue factor (Murata et al., 2006; Stahl et al., 2009).
Renal vascular damage is a prominent feature of HUS, and kidney failure is one of the most serious sequelae during EHEC infection (reviewed in Obrig and Karpman (2012)). A plausible hypothesis is that renal pathology results from pro-thrombotic damage to Gb3-expressing microvascular endothelium of the kidney by circulating Stx, in particular Stx2 (Obrig and Karpman, 2012). Indeed, Stx has been detected in kidney tissue from patients with HUS (Uchida et al., 1999; Chaisri et al., 2001). A possible reason the kidney is specifically targeted may be an elevated Gb3 density in that tissue, because cultured human renal microvascular endothelial cells express 50-fold more Gb3 than human umbilical vein endothelial cells (Obrig et al., 1993). Human glomerular podocytes, mesangial, as well as several other kidney cell types also express Gb3 (reviewed in Obrig and Karpman (2012)). Therefore in addition to the endothelium, other tissues in the kidney may be subject to damage by Stx.
A second clinically important aspect of HUS is the potential development of neurological symptoms. These can result in epileptic seizures, alterations of consciousness, and paresis (Nathanson et al., 2010). Stx2 is able to induce hindlimb paralysis when administered to mice by intraperitoneal injection, suggesting that Stx is directly responsible for some aspects of neurologic disease in EHEC infection (Obata et al., 2008). Consistent with this notion, Stx2 was shown in vivo to act on Gb3-expressing neurons, and treatment of murine brain slices ex vivo resulted in increased Ca2+ transients by astrocytes in response to electric field stimulation.
Complement activation contributes to the vascular damage associated with over 50% of cases of ‘atypical’ HUS, i.e. HUS not associated with diarrheal (e.g. STEC) disease (Besbas et al., 2006; Loirat and Fremeaux-Bacchi, 2011) and may also play a role in Shiga toxin- (diarrhea-)associated HUS (‘D+HUS’) (reviewed in Noris et al. (2012)). C3 deposits on human microvascular endothelial cells that have been treated with Stx1 and perfused with human serum (Morigi et al., 2011). When these cells are perfused with whole blood, Stx1 pretreatment is associated with an increase in cell surface thrombi, an event that is blocked by the complement inhibitor sCR1. Treatment of human serum with Stx2 results in the formation of sC5b-9, indicating that Stx2 is also able to activate complement (Orth et al., 2009). A complement component 3a receptor antagonist diminishes glomerular fibrin deposition and platelet clumps when administered to mice prior to challenge with Stx2 and LPS (Morigi et al., 2011) and mice deficient in factor B of the alternative complement pathway did not suffer renal impairment upon Stx2/LPS challenge. Children with documented or suspected D+HUS had perturbations in serum levels of complement components, e.g. decreased levels of C3 or increased levels of C3b, Ba, Bb, and/or sC5b-9 (Cameron and Vick, 1973; Kaplan et al., 1973; Monnens et al., 1974, 1980; Robson et al., 1992; Thurman et al., 2009; Lapeyraque et al., 2011). One study demonstrated that increased plasma levels of C3a and sC5b-9 during the onset of D+HUS subsequently normalized upon recovery (Stahl et al., 2011). Thus, although the signaling mechanism(s) by which Stxs promote complement activation has not been elucidated, these data suggest that complement activation is associated with D+HUS.
As mentioned above, Stx can induce pro-inflammatory signaling via the RSR, PI3/AKT/mTOR pathways, and apoptotic signaling via the RSR and UPR pathways in vitro. HUS patients manifest elevated urinary and/or serum levels of MCP-1, IL-6, IL-8, and TNF-α (Karpman et al., 1995; Inward et al., 1997; van Setten et al., 1998). PMN counts are elevated during HUS, and together with increased IL-8 levels, are prognostic indicators of severe disease (Fitzpatrick et al., 1992; Robson et al., 1992; Fernandez et al., 2007). Furthermore, apoptotic glomerular and renal tubular cells are present in the kidneys of HUS patients (Karpman et al., 1998; Kaneko et al., 2001; Te Loo et al., 2001a). Thus, it is reasonable to hypothesize that the RSR, PI3/AKT/mTOR, and UPR signaling pathways contribute at least in part to the pathology associated with HUS. However, future studies are required to better characterize the mechanism by which HUS develops, and whether other bacterial factors such as LPS, which is an integral component in one murine Stx model (Keepers et al., 2006), act in concert with Stx to promote severe disease.
Subtilase-like toxin (SubAB)
Although STEC encoding LEE are most often associated with major STEC outbreaks, LEE-negative STEC are also a significant contributor to Shiga toxin-mediated disease including HUS (Bonnet et al., 1998; Paton et al., 1999; Newton et al., 2009; Galli et al., 2010). In 1998 a STEC strain, 98NK2, responsible for an outbreak of D+HUS, was found to lack several virulence factors (including LEE) thought to be important to the onset of systemic disease (HUS) (Paton et al., 1999). However, this strain was found to express a new cytotoxin, Subtilase-like toxin AB (SubAB) (Paton et al., 2004). Most often the gene encoding SubAB has been associated with strains that lack eae and has been found in both Stx-encoding and non-Stx encoding E. coli (Wolfson et al., 2009; Buvens et al., 2010; Galli et al., 2010; Tozzoli et al., 2010). Also most studies of HUS disease by subAB-encoding E. coli have been performed in the context of STEC.
SubAB is a novel AB5 toxin that damages cells by proteolytically cleaving the ER-chaperone and master ER-regulator BiP, also termed GRP78 (Paton et al., 2006). This activity by SubAB has been shown to result in activation of the unfolded protein response in human, African green monkey, and mouse-derived cell lines, as well as after injection into mice (Paton et al., 2006; Wolfson et al., 2008). A role for SubAB in promoting thrombotic sequelae such as HUS is supported by studies showing that SubAB treatment of U937 and HUVECs results in increased tissue factor gene expression as well as increased tissue factor procoagulant activity (Wang et al., 2010). In addition, it has been demonstrated that intraperitoneal administration of SubAB into mice causes TMA with damage occurring to the kidney, liver, spleen, and brain (Wang et al., 2007), hemorrhaging in the small intestine (Furukawa et al., 2011), and severe leukocytosis (Wang et al., 2011). These data support a role for SubAB as an ancillary virulence determinant that may promote severe disease in humans by LEE-negative STEC. However, since HUS disease by subAB encoding E. coli has been addressed only in Stx-producing strains, a specific role for SubAB in virulence has not been established.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


