Fig. 34.1
Structure of photopolymerizable macromolecular precursors: a PEG acrylate derivatives, b polyvinyl alcohol (PVA) derivatives, c dextran methacrylate, and d hyaluronic acid derivatives
34.2.2 Photoclick Reactions for Fast Crosslinking or Ligation
34.2.2.1 Thiol-ene
The radical-mediated thiol-ene reaction has all the desirable features of a click reaction, being highly efficient, simple to execute with no side products and proceeding rapidly to give the ligated products with high yields. The photoinitiated thiol-ene reaction is now frequently used for photopolymerization to get highly uniform polymer networks. Thiol-ene photopolymerized networks have a tremendous advantage over networks through traditional photopolymerization in that they form rapidly and quantitatively under ambient atmospheric conditions to yield nearly ideal, uniform polymer networks through a controllable combination of step-growth and chain-growth [5].
In the ideal purely step-growth thiol-ene reaction, the net reaction is simply the combination of the thiol and ene functional groups (Fig. 34.2). For a given thiol, electron-rich enes polymerize more rapidly than electron-poor enes and 1,2-substituted internal enes gave reduced rates and conversions. The original photo induced thiol-ene photopolymerizations utilized abstraction-type photoinitiators such as benzophenone which at the excited triplet state is able to abstract a hydrogen from the thiol and thus initiate the polymerization. Visible and UV-light-sensitive type I initiators with excellent photoinitiation efficiency are better choices owing to improved initiation efficiency. By choosing appropriate excitation wavelength and monomer combinations, initiator-free thiol-ene photopolymerization reactions have been realized. The initiatorless photopolymerizations are very useful to polymerize thick samples without the generation of any colored or volatile by-products.

Fig. 34.2
The thiol-ene reaction
34.2.2.2 Thiol-yne
The first discovery of radical addition of thiols to alkynes can be dated back to eighty years ago, but only in recent years, the reaction was rediscovered as a click process for the facile preparation of multifunctional polymer structures (Fig. 34.3). It was then considered as a sound candidate for replacing or complementing other popular click reactions such as the thiol-ene coupling and the Cu-catalyzed azide–alkyne cycloaddition. The photoinitiation capability of the thiol-yne coupling is successfully exploited for a number of emerging applications including that for both spatially and temporally controlled functionalization of surfaces [6]. Since the vinyl radicals upon photoinitiation are formed in an irreversible manner and can abstract a hydrogen atom from the thiol reagent more rapidly than their alkyl counterparts, the thiol-yne coupling is most efficient when it is carried out with equimolar amounts of thiol and alkyne. It is noteworthy that highly diluted reaction system might result in appreciable side reactions such as dimerization of sulfanyl radicals to give the corresponding disulfide.
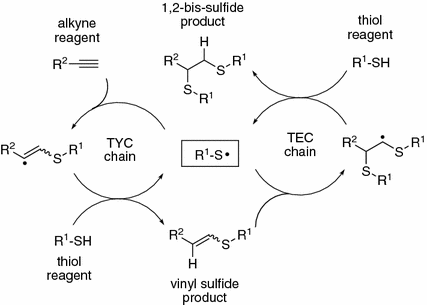
Fig. 34.3
Mechanism of thiol-yne coupling (TYC) and thiol-ene coupling (TEC) radical chains
34.2.2.3 Tetrazole-Alkene
In the late 1960s, Huisgen and co-workers described the first photoinduced 1,3-dipolar cycloaddition reaction between 2,5-diphenyltetrazole and methyl crotonate in benzene at 20 °C. Not until 2007, was the reaction revisited and developed as a “photoclick” reaction which was able to be initiated with a hand-held UV lamp to drive the reaction to completion within two hours [7]. The optimized “photoclick” reaction between tetrazole and alkenes have revealed full water compatibility, high photoactivation quantum yield, tunable photoactivation wavelength and broad substrate scope. Moreover, the formation of fluorescent cycloadducts was claimed as an added benefit in its applications as a spatiotemporally controllable tool to visualize proteins in live cells [8]. The fast reaction kinetics (up to 11.0 M−1 s−1) under mild photoirradiation conditions and the fluorescent cycloadducts of this type of photoclick reactions make them attractive for the engineering of photomanipulatable biomaterials.
34.2.3 Photoisomerization for Reversible Regulation
The ability to reversibly manipulate the physical and chemical properties of a material with an external stimulus forms the basis of stimuli-responsive systems. The application of photochromism to photoresponsive systems has led to the development of new tailored smart materials for photonics and biomedical fields. Within a polymeric matrix, photochromic isomerizations can be stimulated by light to reversibly alter the physical and chemical properties of a material such as phase, shape, self-assembly, size, and fluorescence [9]. Many of the ideas for such photoresponsive systems have been inspired by nature which has evolved many complex biological systems able to exploit light as an external source of energy and information. The underlying principles behind photoresponsive behavior are the photoisomerization of chemical moieties such as azobenzene, spiropyran, and diarylethene.
34.2.3.1 Azobenzene
Azobenzene and its derivatives have been widely used as molecular switches in the area of polymer materials, surface modifications, protein probes, and molecular machines due to their reversible transformations of the trans and cis form upon irradiation with UV or visible light (Fig. 34.4). Photoinduced isomerism of azobenzene also proceeds with large structural change as reflected in the dipole moment and change in geometry. Quantum yields of the transformation are generally high for the isomerization of azobenzene, and there are no competing reactions of significance [10]. Generally, the trans-azobenzene is more stable than the cis form with a trans–cis transform activation barrier of 10–12 kJ mol−1 in solution. It is also worth mentioning that continuous irradiation of trans-azobenzene with either 313 or 436 nm radiation results in a photostationary state comprised of ~20 or ~90 % of trans-azobenzene, respectively.
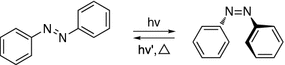
Fig. 34.4
Reversible isomerization of azobenzene
Different ring substitutions have great impact on the absorption, emission, and photochemical property of the azobenzenes. For example, the electron denoting group at the ortho or para position of the phenyl group can dramatically red-shifted the spectrum. Though the red-shifted azobenzenes with increased dipolar character normally exhibit faster thermal relaxation processes (i.e., short-lived cis species), absorption at longer wavelengths with long-lived cis species were found to be possible by modifying delocalization and steric effects. Substituents also can make the cis form more thermodynamically stable than the trans form [11]. With these advances in the development of azobenzene switches, effective control of a large variety of biomaterials for translation medicine can be realized.
34.2.3.2 Spiropyran
Spiropyrans are widely presented in modern scientific literature due to their unique photochromism properties. Upon UV irradiation, molecules with spiropyran moiety are able to be transformed into the MC forms, which reverse transformation can be realized both thermally and photochemically (by irradiation with visible light) (Fig. 34.5). The ring open form is characterized by a significant charge separation, which is usually depicted as zwitter-ionic structure. In general, the ring-closed spiropyran form has absorption in visible range, whereas the merocyanine form is dominated by a strong absorption in the visible range [12]. This photochrome moiety has been subjected to various biological environments like RNA, DNA, and proteins for spatial-temporal control over the bimolecular functions.
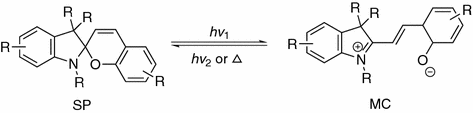
Fig. 34.5
Reversible isomerization of spiropyrans
The most widely studied spiropyran is the 1′,3′,3′-trimethyl-6-nitrospyro [2H-1-benzopyran-2,2-indoline] having the nitro group in the 6 position of the chromene moiety. The nitro-substituted spiropyran obsesses a high quantum yield of photochromic reaction. And the nitro group strongly enhances the intersystem crossing, thereby changing the reaction pathway in photochromism and shift the equilibrium toward the open form. In addition, this kind of spiropyran has been found to undergo hydrolysis in aqueous solutions and lose switching efficiency in the presence of a biomolecule [13].
34.2.4 Photocleavage Reactions for Photodegradation
Unlike the photoclick reactions, the photocleavage reactions are widely used in the photomanipulable materials for photodegradation of the materials or the release of bioactive molecules to modulate the biomechanical and biochemical properties of the biomaterials. One of the most commonly used photolabile groups is the photocleavable o-nitrobenzyl group with all its derivatives [14]. The mechanism of the photolysis of nitrobenzyl (NB) or nitrophenylethyl (NPE) caged compounds has been established to be an intramolecular H-abstraction by the excited nitro group, which is followed by the formation of the aci-nitro form and the rearrangement to the nitroso derivatives [15] (Fig. 34.6). One disadvantage of the ortho-nitrobenzyl group is that upon photolysis a nitrosoaldehyde is formed which can be harmful in a biological systems. The NPE group can be deprotected faster and results in the formation of a nitrosoketone. Excitation wavelength and kinetics of the cleavage process can be fine tuned by the methoxy substituents. Very recently, the nitrodibenzofurane chromophore has been reported to have a significant high extinction coefficient and quantum yield in the near UV region and also suitable for two-photon activation. Other photolabile groups containing coumarin-4-ylmethyl and its derivatives, p-hydroxyphenacyl group, 7-nitroindolinyl-based group have also got increased interest in the photomanipulatable materials.
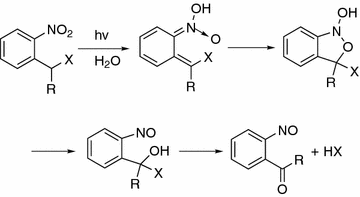
Fig. 34.6
Mechanism on the photolysis of o-nitrobenzyl groups (for NB, R = H and for NPE, R = CH3)
34.3 Photomanipulatable Hydrogel
Hydrogels have emerged as promising synthetic analogs of extracellular matrixes for basic cell studies as well as promising biomaterials for regenerative medicine applications [16]. The artificial matrixes based on hydrogels have critical advantages over natural networks in that bioactive functionalities, such as cell adhesive sequences and growth factors, can be incorporated in precise densities without influence on the substrate mechanical properties. Therefore, hydrogels have become a class of biomaterial scaffolds that have been widely used in complex device fabrication, drug release, and tissue engineering.
Tissue engineering employs cells seeded on or in three-dimensional natural or synthetic scaffolds for tissue regeneration or replacement. The static hydrogel systems for tissue engineering have been well developed so far, while the design of dynamic tunable and biomimetic hydrogel systems for cell scaffold just emerged at the beginning of this century. One of the challenges in the engineering of manipulatable hydrogels is to directly control the gelation process under physiological conditions with no harm to the cell encapsulated in the hydrogel system. Due to the spatial and temporal resolution of photoregulation, light irradiation becomes an ideal external stimuli to manipulate the properties and functions of biomaterials including hydrogels. The engineering of photomanipulatable hydrogels is of great research interests as well as potential applications in translational medicine.
Photolabile moieties and photosensitive reactions provide a means to spatially and temporally manipulate mechanical and biochemical microenvironment of the hydrogels. Biocompatible ways to build the photomanipulatable hydrogels have been developed and their applications as drug release systems, and cell-culture matrices in translational medicine have been demonstrated. Photopolymerizable and photodegradable hydrogels are the most widely applied photomanipulatable biomaterials in the field of tissue engineering for the engineering of tissues such as bone, cartilage, and liver [17]. The spatial and temporal control afforded by photoinitiated polymerization has promoted the development of injectable materials that can deliver cells and growth factors, as well as for the fabrication of scaffolding with complex structures.
34.3.1 Photopolymerizable Hydrogels
Photopolymerized hydrogels have been investigated for a number of tissue engineering applications because the in situ formation of these materials provides a minimally invasive manner compared with other methods such as injection. Quite a lot of hydrogel materials formed via photopolymerization processes mild enough to be carried out in the presence of living cells. The biocompatibility and cellular toxicity of the photopolymerized hydrogels have been widely explored. The UV or visible light used and different kinds of photoinitiators were examined by different groups with different cell lines [18]. With low irradiation intensity and photoinitiator concentrations [≤0.01 %(w/w)], cell survival and cell proliferation were not impaired in general. Different cell types may have variable responses to the same photoinitiator, which was indicated by variable correlation between cellular proliferation rate and increased cytotoxicity of the photoinitiator [19]. The use of photoinitiator-free photopolymerization technique for hydrogel preparation has also been developed [20].
With advances in synthetic chemistry, multifunctional monomers and macromers have been synthesized for photopolymerization to form functional hydrogels. Photoreactive groups, such as acrylates and methacrylates, were introduced into the macromers to give photocrosslinkable synthetic polymers including polyanhydrides [21], poly(ethylene glycol) [22], poly(propylene fumarates) [23], poly(α-hydroxy esters) [24], poly(vinyl alcohol) [25], poly(β-amino esters) [26] etc. Natural polymers, such as collagen [27, 28] and polysaccharides [29–32], have also been modified with photoreactive groups including methacrylates, benzophenone and styrene to be further photocrosslinked. These photopolymerizable synthetic or natural polymers have been developed as degradable biomaterials for tissue engineering applications [33]. We will give some representative or recent examples of each type of photopolymerizable hydrogels for tissue engineering applications after those being reviewed elsewhere [4].
Photoencapsulation of chondrocytes into the polymeric networks was reported [34]. An in vitro analysis of bovine and ovine chondrocytes photoencapsulated in a poly(ethylene oxide)-dimethacrylate and poly(ethylene glycol) semi-interpenetrating network showed that the chondrocyte survived well after one day’s encapsulation and continued to grow in the following four weeks. Biochemical analysis indicated significant increase on the proteoglycan and collagen contents over two weeks’ static incubation, indicating good biocompatibility and efficacy of the photopolymerizable hydrogels for the encapsulation of chondrocyte cell with low-intensity UVA light. In another work using the photoencapsulation of osteoblasts in injectable RGD-modified PEG hydrogels for bone tissue engineering [35], it was reported that a majority of the osteoblasts survived the photoencapsulation process when gels were formed with 10 % macromer. However, a decrease in osteoblast viability of ∼25 and 38 % was observed for the encapsulated cells after 1 day of in vitro culture when the macromer concentration was increased to 20 and 30 %, respectively.
The photopolymerized poly(ethylene glycol)-based hydrogel has been demonstrated as regenerative medicine for in vitro chondrogenesis of the bone marrow-derived mesenchymal stem cells (MSCs) [36]. The MSCs from skeletally mature goats encapsulated in the hydrogel were cultured with or without transforming growth factor β 1(TGF). It turned out that the encapsulated MSCs in the chondrogenic medium with TGF proliferated, while the glycosaminoglycan and total collagen content of the hydrogels increased to 3.5 % dry weight and 5.0 % dry weight in 6 weeks. The results demonstrated the potential of the photopolymerizing hydrogel to encapsulate MSCs and induce the formation of cartilage-like tissue for stem-cell translation medicine.
HA is a naturally derived polysaccharide. The monomer of HA is composed of D-glucuronic acid and N-acetyl-D-glucosamine. It is attractive for biomaterial applications, because it can be modified with various functional groups which make covalent crosslinking reactions possible and it is degradable upon treatment of the enzyme hyaluronidase. Besides, HA has proved to play roles in the promotion of cell motility and proliferation, wound healing, angiogenesis, and the reduction of long-term inflammation [37]. It has been reported that photopolymerized HA-based hydrogels and interpenetrating networks were readily degraded by hyaluronidase and their mechanical properties could be modulated by factors such as molecular weight of HA and concentration of PEG-DA [38]. The incorporation of RGD peptides further allowed modulation of the HA properties from cell nonadhesive to adhesive.
Visible-light crosslinkable hydrogel based on methacrylated glycol chitosan (MeGC) has been presented recently [39]. Three blue light initiators, camphorquinone (CQ), fluorescein (FR), and riboflavin (RF), were tested in the photocrosslinking. A minimal irradiation time of 120 s was required to produce MeGC gels able to encapsulate cells with CQ or FR. Prolonged irradiation up to 600 s improved the mechanical strength of CQ- or FR-initiated gels but drastically reduced viability by 5 and 25 % for chondrocyte encapsulated in the photocrosslinked gels initiated by CQ and FR, respectively. Increasing the irradiation time up to 300 s significantly improved the compressive modulus of the RF-initiated MeGC gels without reducing cell viability. RF-photoinitiated hydrogels supported proliferation of encapsulated chondrocytes and extracellular matrix deposition. In osteochondral and chondral defect models, the in situ gelling hydrogels using RF was further demonstrated for potential cartilage tissue engineering.
The step-growth thiol-ene photoclick hydrogels have also showed superior properties including cytocompatibility of the reactions and improved hydrogel physical properties. The thiol-ene or thiol-yne reaction involves the sequential propagation of a thiyl radical with an alkyl or vinyl functional group followed by chain transfer of the radical to another thiol to form the network [40, 41]. Thiol-ene click reactions provide an attractive means to fabricate PEG hydrogels with superior gel properties for in situ cell encapsulation, as well as to generate and recover 3D cellular structures for regenerative medicine applications. Compared with the disordered network formed by polymerization of PEG diacrylate analogs, thiol-ene step-growth photopolymerizations have demonstrated the advantage of forming highly ordered polymer networks. McCall and Anseth compared the differences in recovery of bioactive proteins when exposed to similar photoinitiation conditions during thiol-ene versus acrylate polymerizations [40]. After exposure to chain polymerization of acrylates, lysozyme bioactivity was approximately 50 %, after step-growth thiol-ene reaction, lysozyme retained nearly 100 % of its prereaction activity. Bioactive protein recovery was enhanced 1,000-fold in the presence of a thiol-ene reaction, relative to recovery from solutions containing identical primary radical concentrations, but without the thiol-ene components. This showed a potential for this polymerization method in the bioactive macromolecules encapsulation and controlled delivery in vivo.
The step-growth thiol-ene photoclick hydrogels also demonstrated the ability for 3D culture of pancreatic β-cells. Cells encapsulated in the thiol-ene hydrogels formed spherical clusters naturally and were retrieved via rapid chymotrypsin-mediated gel erosion. The recovered cell spheroids released insulin upon glucose treatment, demonstrating the cytocompatibility of thiol-ene hydrogels and the enzymic mechanism of cell spheroids recovery [42]. Hydrogels synthesized by a step-growth reaction mechanism via thiol-norbornene photopolymerization have also been reported to be with rapid gelation time and high cell viability for cell encapsulated [43]. It has also been demonstrated that the hydrogel combined the advantages of ideal, homogeneous polymer network formation, facile incorporation of peptides without post-synthetic modification, and spatial and temporal control over the network evolution into a single system with enzyme- and cell-responsive characteristics. The high degree of spatial and temporal control over gelation, combined with robust material properties, makes step-growth thiol-ene photoclick hydrogels hydrogels an excellent tool for a variety of medical applications.
In 2006, Engler et al. demonstrated that microenvironments appear important in stem-cell lineage specification. Soft matrices that mimic brain are neurogenic, stiffer matrices that mimic muscle are myogenic, and comparatively rigid matrices that mimic collagenous bone prove osteogenic [44]. As for naive MSCs, during the initial week in culture, reprogramming of these lineages is possible with addition of soluble induction factors, but after several weeks in culture, the cells commit to the lineage specified by matrix elasticity, consistent with the elasticity-insensitive commitment of differentiated cell types. The results have significant implications for understanding physical effects of the in vivo microenvironment and also for therapeutic uses of stem cells, but it can be difficult to adequately characterize or control with soft tissues or static in vitro models. Hence, the photo enhanced hydrogel provides a unique and dynamic way for studying relationships of the stem-cell differentiation and the microenvironment changes.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


