Endometrial Hyperplasia without Atypia and EIN
The Spectrum of Non-Atypical Endometrial Hyperplasias
Disordered Proliferative Endometrium: a Prelude to Non-Atypical Hyperplasia
Non-Atypical Hyperplasia with Superimposed Progestin Effect
Withdrawal Shedding Following Non-Atypical Hyperplasia
Cancer Outcomes in Women with EIN
Molecular Etiology and Natural History
Specific EIN Diagnostic Criteria
Common EIN Diagnostic Problems
Special Studies for EIN Diagnosis
Introduction and Terminology
The meaningful resolution of endometrial hyperplasias, a mixed group of diseases, has challenged pathologists for decades. Long envisioned as a continuous spectrum of morphologic changes of increasing severity, in reality they encompass only two discrete disease states, which can and should be diagnosed independently of carcinoma (Table 17.1). This consensus has now been endorsed by the Clinical Practice Committee of the Society of Gynecologic Oncologists1 and the World Health Organization (WHO).2 Endometrial hyperplasia without atypia is a functionally normal endometrium which has developed changes in response to an abnormal hormonal environment, usually unopposed estrogens. EIN, endometrial (or, endometrioid) intraepithelial neoplasia, also known as atypical hyperplasia, is a monoclonal proliferation of mutated glands that increases the likelihood of carcinoma through malignant transformation of its component cells. We have used the terms non-atypical hyperplasia and EIN throughout this chapter, corresponding to the respective WHO 20142 categories of hyperplasia without atypia, and Endometrioid Intraepithelial Neoplasia. The supportive evidence base for EIN has already been published extensively,3,4 and has been clinically implemented since 2001 as part of a two-class system.5
Table 17.1
Diagnostic Entities and Terminology (WHO, 2014)
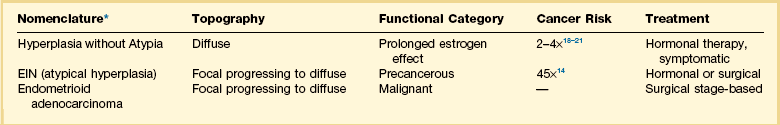
*In WHO 2014 terminology, EIN and atypical hyperplasia are alternative, synonomous, terms.
All examples of endometrial hyperplasia are abnormal in having too many glands, with distinction between non-atypical endometrial hyperplasia and EIN based upon the character and architectural context of those cytologic changes that are present. The intrinsic plasticity of the endometrium, and its dynamic response over time to a large number of internal and external factors, contributes a great deal of variation to the appearances of individual lesions between patients. It has been the examination of many examples of functionally classified disease that has provided a new evidence base for their diagnosis.6 As several of the most informative diagnostic criteria available today were unknown at the time of the formulation of the 1993 four-class WHO system (simple non-atypical, simple atypical, complex non-atypical, complex atypical),7 the current 2014 version is a replacement for, rather than condensation of, the prior system.
Two Diseases
Endometrial hyperplasia is a nonspecific histopathologic term indicating an abundance of glands and increase in tissue bulk (Figure 17.1). Historically, it came to encompass a bewildering array of lesions that were further subdivided by morphology and suspected to include precancers as one of its subsets. New possibilities emerged in the last two decades, as molecular tools were applied to the problem of precancers. It became possible to query growth patterns (polyclonal, monoclonal), develop markers to highlight lesions against the backdrop of their tissue of origin, and track cell lineages between premalignant and malignant phases. As a result of these changes, a National Institutes of Health workshop convened in 2004 to define features of epithelial precancers that are shared across multiple tissues.8 These included: (1) the lesion is different from normal tissues,9 (2) the lesion is different from cancer,10,11 (3) the lesion can be diagnosed,12 (4) there is lineage continuity between cells of the precancer and cancer,6,11 and (5) patients with the lesion have increased rates of cancer.13,14 All of these evidence-based criteria had already been met by EIN, as indicated by citations inserted into the preceding numbered list. Field effects, such as the general response of a tissue to hormonal, irradiation, or infectious factors, do not meet the definition of a precancerous lesion, as these are intrinsically normal tissues reacting to external influences (failed criterion 1) and it is not possible to identify specific cells that will become cancer (failed criterion 4).
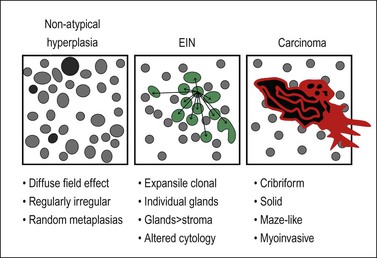
Figure 17.1 General features that distinguish EIN from non-atypical hyperplasia and carcinoma. (For carcinoma, see also Figure 18.12.)
The neoplastic character of EIN, originally documented by nonrandom X chromosome inactivation,15 has since been shown to bear clonal abnormalities of PTEN,10 PAX2,16 KRAS2,17 and altered microsatellites.6,11 In the case of those markers which can be studied in situ by immunohistochemistry, PTEN and PAX2, staining of informative lesions has been useful in defining lesion distribution.16 Most importantly, the localized clonal character of premalignant lesions affords an opportunity to compare lesional with background cytology in a robust internally controlled assessment of cytologic alteration. Such a standard holds across shifting hormonal states, and between patients, in whom the lesions themselves may have a variety of absolute cytologies. Additionally, large-scale ‘topographic’ features are informative in distinguishing localized precancers from the diffuse field effects of hormonal change. Morphometry of biologically defined precancers (monoclonal, in a continuous lineage to cancer) has contributed precise definition of gland crowding in precancers. These simple insights provide a new evidence base for revision of precancer diagnostic criteria as presented below.
Prolonged estrogen exposure unmitigated by opposing progestins and the appearance of an abnormal endometrial histology have long been associated with an increased risk for endometrioid (type I) endometrial adenocarcinoma. The risk, which is two- to four-fold increased,18–20 has been calculated from large epidemiologic studies of cancer outcomes in women of known hormonal exposure. The histologic appearance of the endometrium modified by unopposed estrogens is initially dependent on dose and duration of exposure, but uniform throughout the field. These can be heavily modified when the estrogens decline or are subsequently countered by progestins. The resultant spectrum of histologies, which we have described as a dynamic sequence of change over time, is discussed below as non-atypical endometrial hyperplasia.
The Spectrum of Non-Atypical Endometrial Hyperplasias
Non-atypical endometrial hyperplasia is a spectrum of hormonally induced pan-endometrial changes, characterized by increased gland density and variation in gland size and shape. Glands may branch slightly. Cytologic change, when it occurs, is most often metaplastic and distributed in a scattered, non-geographic or random fashion. Areas of stromal breakdown and epithelial collapse may be present. This appearance changes dynamically as a function of interval and amount of unopposed estrogens,21 may persist in altered form after the exposure ends, and can be modified by superimposed secondary hormonal effects such as by progestins.22
Non-atypical hyperplasia of the endometrium has many synonyms including simple or complex non-atypical hyperplasia,23 endometrial hyperplasia,4 and benign endometrial hyperplasia.22 It is related to disordered proliferative and anovulatory endometrium, which are lesser changes seen with shorter estrogen exposures (see Chapter 15).
Clinical Features
The two- to four-fold increased risk of endometrioid endometrial cancer in patients with non-atypical hyperplasia is referable to the extent of estrogen exposure.18–21 Clinical treatment is focused on symptomatic relief, and identification and control of estrogens. This can be a difficult task, especially when the underlying condition is a chronic state, such as obesity or polycystic ovarian syndrome.
Etiology
Longer intervals of estrogen stimulation cause an even greater exaggeration of the proliferative phase, with an increasingly irregular distribution of individually variable endometrial glands, which are known as non-atypical hyperplasia. Looked at in this way, disordered proliferative endometrium and non-atypical hyperplasia may be regarded as a physiologic response of a normal endometrium to prolonged and unopposed estrogen stimulation. Microinfarcts and estrogen withdrawal are responsible for symptomatic bleeding.24,25 Both mechanisms may be effective at different times in patients with non-atypical hyperplasia. Patchy stromal breakdown secondary to estrogen-induced microthrombi can produce intermittent spotting.
The pharmacologic administration of estrogen, or estrogenic compounds, can cause a persistent proliferative state in the postmenopausal patient.26 As with anovulation, the root cause is the systemic hormonal imbalance, and the endometrial response a frequent source of symptomatic bleeding.
Gross Features
Initial changes, seen after only 3 weeks of unopposed estrogens (1 week beyond expected ovulation date), are subtle. The endometrium may be moderately but uniformly thickened with a smooth surface lining. Scattered microcysts, although often present, are not apparent on gross examination. With further estrogen exposure, the uterus may enlarge and the endometrium often is irregularly thickened, pale to tan, and sometimes sessile polypoid (Figure 17.2). Endometrial curettage in a woman who has non-atypical hyperplasia is likely to generate abundant curettings that require two or more cassettes for adequate processing. Foci of hemorrhage may be present in the areas of breakdown. Ultrasound may help identify the thickened endometrium.27
Microscopic Features
The histologic changes of non-atypical hyperplasia are conceptually and morphologically well represented as a disease spectrum related to unopposed estrogens. The morphologic features are characteristically diffuse and widespread, as would be expected for a tissue responding uniformly to an underlying systemic hormonal cause. Individual examples differ by time line and amount of estrogen exposure, and may be further modified by degenerative changes or other sex hormones such as progestins (Table 17.2). The range of possible histologic changes is thus broad, with few specimens demonstrating them all. Aside from providing some measure of the patient’s hormonal state at the time of biopsy, discrimination among the various histologic subtypes of non-atypical hyperplasia discussed below is of little immediate clinical importance. Much more critical is that non-atypical hyperplasia is not confused with EIN (Figure 17.1). These are not mutually exclusive in the individual patient, however, as non-atypical hyperplasia is a common comorbidity in women with EIN. The sections below provide a functional framework to describe the histologic varieties of commonly encountered non-atypical endometrial hyperplasia.
Table 17.2
Histologic Features of Non-Atypical Hyperplasia (Not All Are Present in Every Case)
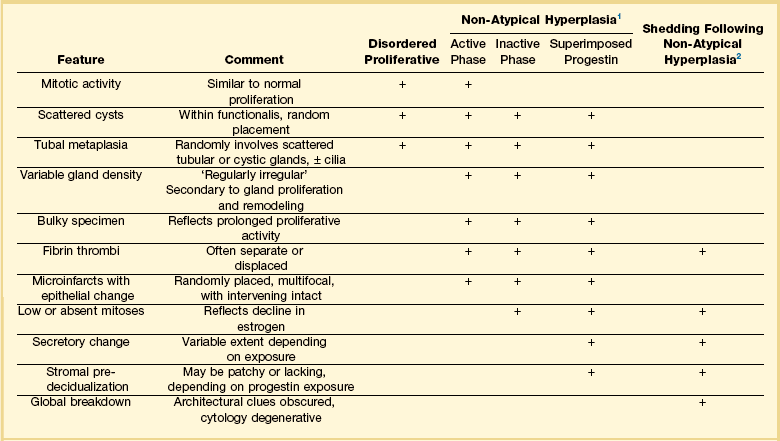
1Diagnosis is ‘non-atypical hyperplasia.’ Subphases need not be specified.
2Indistinguishable from menstrual endometrium, with or without confirmation of ovulation. If present, fibrin thrombi can be described as a feature consistent with a hormonally abnormal antecedent cycle.
Disordered Proliferative Endometrium: a Prelude to Non-Atypical Hyperplasia
Disordered proliferative endometrium is an exaggeration of the normal proliferative phase cause by failed ovulation or minor prolongation of estrogen stimulation. The pathognomonic feature is cystic changes of individual glands distributed randomly throughout the entire hormonally responsive region of the endometrium (superficial functionalis) (Figure 17.3). The overall ratio of glands to stroma is not significantly increased from that of a normal proliferative phase, and mitotically active glands are proliferative with a simple but often pseudostratified epithelium (Figure 17.4). The stroma is usually dense, cellular, and abundant, thus separating the glands. Gland density is low. Some background tubular glands are slightly irregular and minimal budding and branching is commonly seen.
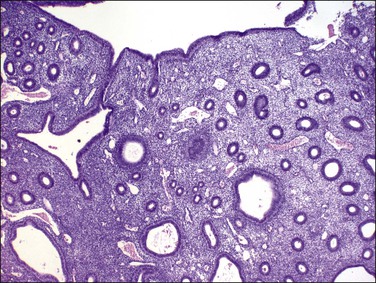
Figure 17.3 Disordered proliferative endometrium demonstrating glands at low density with scattered cyst formation. Placement of glands is quite regular, and branching is slight, indicating a minimal extent of remodeling.
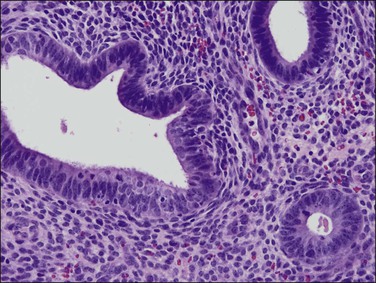
Figure 17.4 Cytology of disordered proliferative glands resembles that of normal proliferative endometrium, surrounded by a dense stroma.
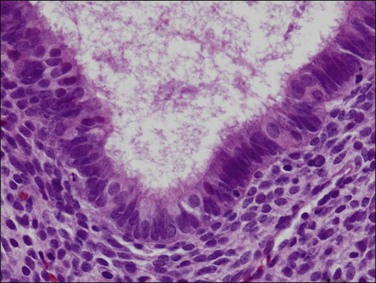
Ciliated cell change (tubal metaplasia) of endometrial glandular cells is common, reflecting estrogen’s pivotal role in the process (Figure 17.5). Characteristically, glands affected by tubal differentiation are randomly interspersed among proliferative glands, and they may demonstrate tubular, branching, or cystic architecture.
Non-Atypical Hyperplasia
Non-atypical hyperplasia develops from disordered proliferative endometrium under the continued influence of unopposed estrogens. The entire endometrial compartment contains variable gland densities caused by remodeling of stroma and glands to the extent that in some areas the gland to stroma ratio exceeds 1. It is the change in gland density that offsets non-atypical hyperplasia from disordered proliferative endometrium. Individual glands may be tubular, cystic, or branching, and these forms are commingled throughout. On a large scale the endometrium appears uniformly affected; however, at medium magnification local admixtures of individually variable glands present quite differing appearances between separate microscopic fields. This combination of low-magnification uniformity, made up of variable medium magnification fields, can be described as ‘regularly irregular’ (Figure 17.6).
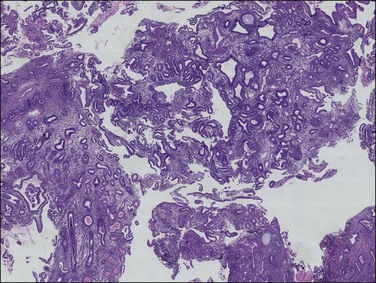
Figure 17.6 Macroscopic view of non-atypical hyperplasia showing randomly distributed cysts and variable gland spacing across multiple fragments. This presents a ‘regularly irregular’ pattern, in which there is a broad symmetry among fragments at very low magnification, which disappears at smaller scales where each field looks different.
The cytology of non-atypical hyperplasia does not change between architecturally crowded and uncrowded areas. This reflects the systemic hormonal etiology of the process, which similarly exposes the entire endometrium, and allows its distinction from EIN. Absolute cytologic presentation may change over time with the evolving hormonal state of the patient, and superimposition of local factors such as breakdown and repair. During the established phase of active estrogen exposure glands are proliferative and interposed tubal metaplasia is common (Figures 17.7 and 17.8).
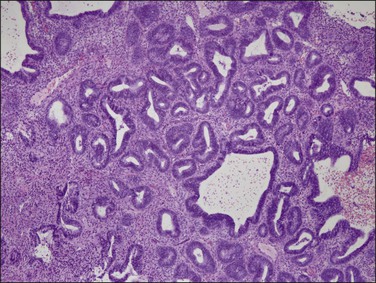
Figure 17.7 Non-atypical hyperplasia with randomly interspersed cystic and tubular glands. The entire endometrial compartment is involved, but local remodeling of glands with stroma creates regional heterogeneity of gland density. Glands are tubular, cystic, and branching.
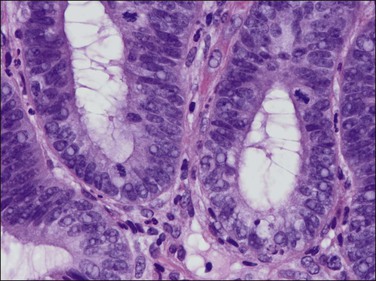
Figure 17.8 Non-atypical endometrial hyperplasia, detail. Proliferative glands indicate an active phase of estrogen stimulation.
Sometime after initiation of cystic gland dilatation the endothelial lining of ectatic superficial endometrial vessels may be damaged and occlusive luminal fibrin thrombi form (Figure 17.9). Unopposed estrogen states are the most common setting in which fibrin thrombi are seen in the intact endometrial functionalis.24 Fibrin thrombi are rarely seen in architecturally normal late secretory endometrium, and there is no evidence that vascular thrombosis is a primary mechanism of cyclical synchronized menstrual shedding. Thrombi are often intimately associated with discrete areas of surrounding stromal breakdown, which has been interpreted as either a cause or effect of the vascular lesion. Whatever the sequence and mechanism of events, the two are linked in non-atypical hyperplasias, and are responsible for patchy non-synchronous endometrial breakdown and resultant symptoms of spotting and intermenstrual bleeding. Collapse of intervening broken down stroma may lead to close apposition of endometrial glands, degenerative epithelial changes (Figure 17.10), and dislodgement of vascular thrombi from their tissue context. It is the close association of these epithelial changes with stromal breakdown that permits their distinction from EIN.
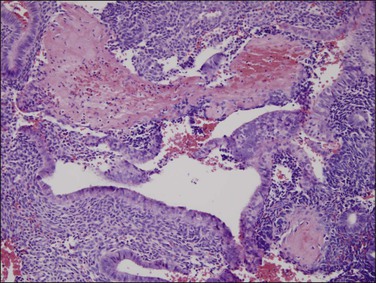
Figure 17.9 Fibrin thrombi can be seen in superficial blood vessels of non-atypical hyperplasia, causing local breakdown.
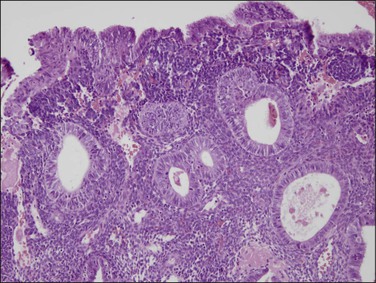
Figure 17.10 Stromal breakdown in non-atypical hyperplasia alters the cytology of the adjacent epithelium, and may lead to gland displacement.
Estrogen production from persistent follicles or by peripheral conversion following the menopause is inconstant. When the estrogen level declines slowly, massive breakdown does not occur and the glands lose mitotic activity and enter an inactive phase. These endometria retain the architectural features of a bulky endometrium with altered gland architecture, but the glands lack mitotic activity (Figures 17.11 and 17.12) and may become karyorrhectic. With prolonged low estrogen levels, endometrial bulk declines toward an atrophic pattern, sometimes with cysts.
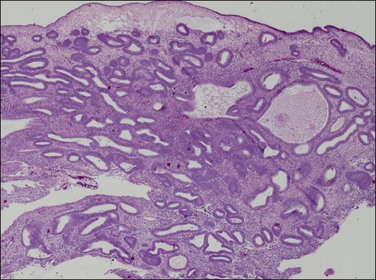
Figure 17.11 Non-atypical hyperplasia, inactive phase. Irregular and cystic glands are present at high density.
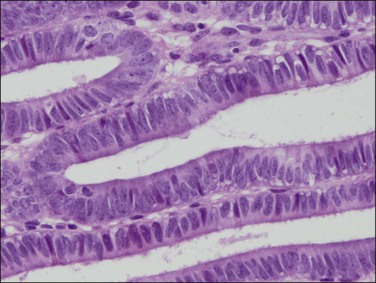
Non-Atypical Hyperplasia with Superimposed Progestin Effect
Superimposition of endogenous or exogenous progestins upon non-atypical hyperplasia shuts down mitotic activity, and may initiate secretory change. The most common endogenous progesterone source is delayed ovulation in a perimenopausal woman, where the corpus luteum is typically unable to elaborate normal quantities and duration of progesterone (Figures 17.13 and 17.14). The process is widespread throughout the endometrial compartment, although the secretory response of glands may be patchy or irregular when progestin levels are subphysiologic. More profound progestin effects, including secretory exhaustion and extensive stromal pseudodecidualization, can be seen with exogenous hormonal therapy (Figure 17.15).
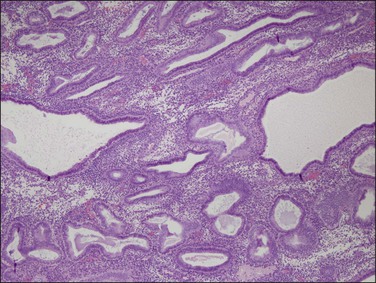
Figure 17.13 Delayed ovulation following protracted estrogen stimulation superimposes secretory change upon pre-existing cystic architecture.
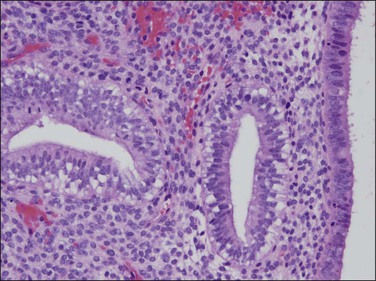
Figure 17.14 Detail of delayed ovulation in non-atypical endometrial hyperplasia showing vacuoles, which indicate progesterone effect. The progesterone is of short duration, as mitoses are not yet suppressed.
Withdrawal Shedding Following Non-Atypical Hyperplasia
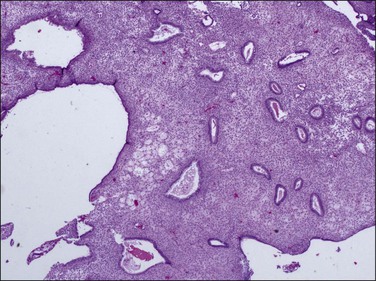
Cessation of estrogenic stimulation, such as occurs systemically upon shutdown or exhaustion of the persistently active ovarian follicle, leads to rapid endometrial-wide stromal breakdown and heavy menses. This occurs through a direct apoptotic effect upon endometrial stromal and epithelial cells, rather than thrombosis-initiated infarction responsible for breakdown during the estrogen-rich period. Architectural features of cysts and irregular gland distribution are increasingly obscured by stromal collapse, eventually yielding a nondescript collection of degenerative individual glands. For these reasons, it can be difficult to confirm in the late stages of shedding whether the preceding cycle was normal or abnormal, or whether a non-atypical hyperplasia was present or not. Fibrin thrombi may remain, but their presence in itself is nonspecific and insufficient for a diagnosis of non-atypical endometrial hyperplasia (Figure 17.16).
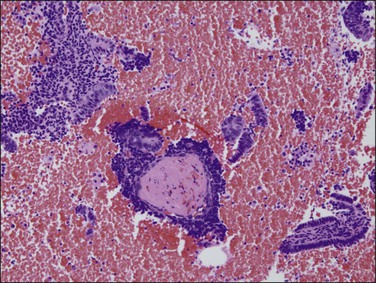
Figure 17.16 Withdrawal shedding of non-atypical hyperplasia is caused by rapid decline of supportive sex hormones. Fibrin thrombi scattered among nondescript stromal aggregates may be the only identifiable residua, as more specific diagnostic features are obscured or ‘erased’ by massive breakdown.
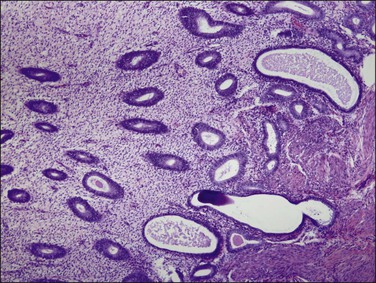
Normal Endometrium
Immediately following menses and at the beginning of the proliferative phase, the endometrial functionalis is thin, with endometrial basalis constituting the majority of the tissue seen. Irregularly spaced and occasionally branching glands of the basalis must be distinguished from non-atypical hyperplasia. The glands of basal endometrium are dark and irregular and may be cystic but they are localized to the endomyometrial junction (Figure 17.17). The basal stroma is dense and cellular. Endometrium sampled between cessation of menses and onset of proliferation is much scantier than that of non-atypical hyperplasias, and there is usually evidence of recent repair along the luminal surface.
Endometrial Polyps
Endometrial polyps (see Chapter 16), especially when fragmented, are commonly misdiagnosed. Within individual polyp fragments, the irregularly distributed and occasionally branching glands with occasional cysts bear some resemblance to non-atypical hyperplasia. Thick-walled blood vessels and fibrous stroma commonly seen in polyps are lacking in non-atypical hyperplasia. Further clues are provided by careful examination of all available fragments. Because polyps are localizing lesions, specimens obtained by undirected biopsy or curettage typically contain commingled native endometrium with a completely different polyp pattern. This is not the case with non-atypical hyperplasia where the entire functionalis is affected.
Postmenopausal Cystic Atrophy
A commonly encountered pattern that may be mistaken for non-atypical hyperplasia is seen during the perimenopausal and postmenopausal periods and is composed of prominent cystically dilated glands in scant, often septated, fibrous stroma. In the past this was one of two patterns often called ‘Swiss cheese endometrium.’ The terms ‘cystic atrophy’ or ‘cystic atrophic endometrium’ accurately describe these appearances and show cuboidal or flattened and inactive cells lining the distended glands. In contrast the epithelium of non-atypical hyperplasia is thicker and may be pseudostratified. Furthermore, the glands in cystic atrophy lack budding and infoldings.



