Overview
In general, most endocrine pathology involves either the overproduction or the underproduction of a hormone. Overproduction of a given hormone may be caused by hyperplasia of the organ that produces the hormone, by a neoplastic process, or by some combination of the two processes. Underproduction of a given hormone, in contrast, may be caused by either destruction of the gland that produces the hormone or by conditions that deprive an endocrine organ of its normal trophic influence.
A working knowledge of the pathways that regulate normal hormone levels helps to interpret the laboratory values in patients being worked up for suspected endocrine disorders. For example, thyroid-releasing hormone (TRH) released by the hypothalamus stimulates thyroid-stimulating hormone (TSH) production by the pituitary gland, which in turn stimulates triiodothyronine (T3) and thyroxine (T4) production by the thyroid gland. T3 and T4 then cause feedback inhibition of pituitary release of TSH. If the patient has a TSH-secreting pituitary adenoma, T4 and T3 levels as well as the TSH will be high; normally, high T3 and T4 levels should cause a low TSH level. Also, remember primary diseases are diseases that originate within the gland in question (e.g., primary hyperthyroidism is due to a defect in the thyroid gland), and secondary diseases represent change in one organ as a result of disease in another organ (e.g., secondary hyperthyroidism may be due to a TSH-secreting pituitary adenoma).
This chapter will discuss diseases of the pituitary gland (hyperpituitarism, hypopituitarism, mass effect as related to pituitary gland lesions, and posterior pituitary gland pathology), diseases of the thyroid gland (goiter, hyperthyroidism, hypothyroidism, thyroiditis, and thyroid neoplasms), diseases of the parathyroid glands (hyperparathyroidism and hypoparathyroidism), diabetes mellitus, diseases of the adrenal glands (hyperadrenalism hypoadrenalism, hyperaldosteronism, and adrenal neoplasms), and multiple endocrine neoplasia (MEN).
Hyperpituitarism
Overview: Overproduction of pituitary gland hormones, usually referring to those derived from the anterior pituitary gland. Causes of hyperpituitarism include adenomas, hyperplasia, and carcinoma.
Epidemiology: Most pituitary adenomas occur during the fourth to sixth decades of life. Adenomas are the most common cause of hyperpituitarism.

Hormone production
- About 30% of pituitary adenomas produce prolactin. The second most common hormone produced by adenomas is growth hormone (GH), followed by adrenocorticotropin hormone (ACTH). Thyroid-stimulating hormone (TSH) is rarely produced by adenomas. Adenomas that secrete combinations of different hormones are referred to as plurihormonal adenomas. Adenomas producing both prolactin and GH are the most common type of such mixed tumors.
- Null cell adenomas are tumors that do not produce a significant amount of hormone, and are the second most common type of adenoma overall.
Important point regarding pituitary adenomas
- Stalk effect: Secretion of all of the anterior pituitary hormones, except prolactin, is stimulated by delivery of releasing hormones, including TRH, gonadotropin-releasing hormone (GnRH), and corticotropin-releasing hormone (CRH), from the hypothalamus via the hypophyseal portal system. Secretion of prolactin, however, is tonically inhibited by the delivery of dopamine via the same portal system. A mass pressing on the stalk will prevent dopamine from reaching the pituitary gland, thus causing increased levels of prolactin without actually producing prolactin. In general, however, the level of prolactin in the “stalk effect” does not equal that produced by a prolactin-secreting adenoma. This stalk effect will, simultaneously, cause inhibition of secretion of the other anterior pituitary hormones.
Overview: The characteristic features of prolactinomas, GH-producing adenomas, ACTH-producing adenomas, and null cell adenomas will be discussed below.
1. Prolactinoma
Clinical presentation: Prolactinomas are diagnosed earlier in females (between 20 and 40 years of age) than in males, because patients present with symptoms of galactorrhea, infertility, and amenorrhea. In males, prolactinomas cause decreased libido and impotence.
Diagnosis: Basal prolactin level of > 200 ng/mL; brain MRI.
Important point regarding prolactinoma: The differential diagnosis of hyperprolactinemia includes pharmacologic and physiologic causes in addition to pituitary lesions. For example, in primary hypothyroidism, both TSH and TRH are elevated. In addition to stimulating release of TSH, TRH causes release of prolactin; thus primary hypothyroidism is a cause of hyperprolactinemia and galactorrhea. Antipsychotic medications that block dopamine are also a common cause of hyperprolactinemia. Other causes include pregnancy.
2. GH-secreting adenomas
Mutation: Often (40%) have a mutation in the GNAS1 gene on chromosome 20q13, which results in abnormal activation of G protein through inhibition of guanosine triphosphate (GTPase) activity and resultant production of cyclic adenosine monophosphate (cAMP).
Clinical presentation of GH-secreting adenoma
- In children, gigantism (if the adenoma is present before the closure of the epiphyseal plates).
- In adults, acromegaly (if the adenoma develops after closure of the epiphyseal plates). Acromegaly is characterized by growth in skin, soft tissue, thyroid gland, heart, liver, and bones of face, hands, and feet. The most classic feature is acral enlargement, or widening of the hands and feet and coarsening of facial features. Patients with acromegaly have persistently elevated levels of GH, which stimulates insulin-like growth factor-1 (IGF-1), causing abnormal glucose tolerance and diabetes mellitus. Patients also have muscle weakness, hypertension, arthritis, osteoporosis, and congestive heart failure.
Diagnosis: Failure to suppress GH level with an oral load of glucose; IGF-1 levels are elevated.
3. ACTH-secreting adenoma
Diagnosis: Elevated levels of plasma ACTH and plasma cortisol. Suppression of cortisol and ACTH secretion with high-dose dexamethasone challenge, but no response to low- dose dexamethasone.
Clinical presentation of ACTH-secreting adenoma: Produce Cushing disease.
Important point: If a patient with an undiagnosed pituitary ACTH-secreting adenoma undergoes adrenalectomy, the pituitary adenoma has aggressive growth due to loss of feedback inhibition. This condition is called Nelson syndrome.
4. Null cell adenomas: Present because of mass effect (see Mass Effect).
Morphology of pituitary adenoma
- Gross: Hormone-producing adenomas can be small (< 1.0 cm). Null cell adenomas are usually much larger at presentation because a larger size is required to produce mass effect. Mass effects produce the symptoms that allow for the tumor to present in a patient.
- Microscopic: Monomorphous proliferation of cells; usually, immunostains are used to identify specific cell types. Adenomas have no reticulin, in contrast to the normal anterior pituitary gland.
- Clinical presentation of pituitary adenoma: A macroadenoma can have symptoms due to mass effect, including headache, visual abnormalities (e.g., bitemporal hemianopsia), and hypopituitarism due to compression and atrophy of the normal portion of the gland. Patients can also have compression of the occulomotor nerve (CN III), trochlear nerve (CN IV), and the abducens nerve (CN VI), with resultant abnormalities of eye movement.
Hypopituitarism
Overview: Underproduction of pituitary gland hormones. Hypopituitarism is noted clinically if there is a loss of 75% or more of the gland.
- Null cell pituitary adenoma: Mechanism is the growth of a nonsecreting tumor that causes destruction of the adjacent normal gland.
- Ischemic injury: For example, in Sheehan syndrome, pregnancy causes enlargement of the pituitary gland due to an increase in the number and size of prolactin-secreting cells. The blood supply of the anterior pituitary is derived from the low-pressure hypothalamic-hypophyseal portal venous system and does not increase despite the increase in the overall size of the anterior pituitary, rendering the anterior pituitary vulnerable to ischemic injury. When blood flow to the anterior pituitary is compromised, as in the case of hypotension associated with obstetrical hemorrhage, necrosis of the gland may occur.
- Pituitary apoplexy
- Basic description: Hemorrhage into the pituitary gland, usually into an adenoma.
- Clinical presentation: Rapid onset headache and diplopia.
- Complications of pituitary apoplexy: Hypopituitarism and diabetes insipidus; potentially cardiovascular collapse and death.
- Basic description: Hemorrhage into the pituitary gland, usually into an adenoma.
- Empty sella syndrome: Condition due to an incompetent diaphragm sella that allows herniation of arachnoid into the sella turcica. The herniated arachnoid presses on the pituitary gland, which causes atrophy of the pituitary gland.
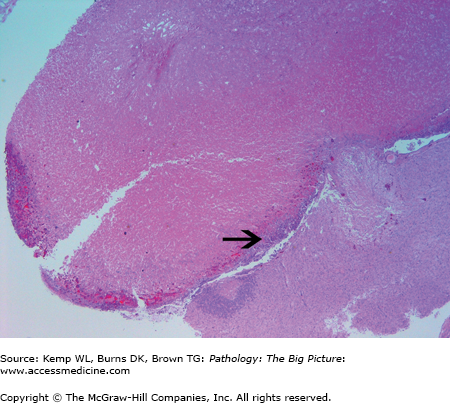
Others causes of hypopituitarism: Surgery, radiation, inflammatory reactions, disseminated intravascular coagulation (DIC), and sickle cell anemia. Necrosis of the anterior pituitary may also occur in the setting of increased intracranial pressure due to compression of the low pressure portal veins supplying the anterior pituitary (Figure 18-2).
- GH deficiency
- In infants: Growth retardation, short stature, and fasting hypoglycemia.
- In adults: Abdominal obesity and reduced strength and exercise capacity.
- In infants: Growth retardation, short stature, and fasting hypoglycemia.
- TSH deficiency: Hypothyroidism (see Hypothyroidism).
- ACTH deficiency: Hypoadrenalism (see Hypoadrenalism).
- ADH deficiency: Diabetes insipidus (see Diabetes insipidus).
Mass Effect
Overview: Mass effect, in general, refers to the effects of a tumor based upon its mass and the involvement of adjacent structures. The pituitary gland is such a small organ surrounded by numerous vital and important structures that mass effects caused by lesions in the pituitary gland are of vital importance in diagnosis. Mass effects are most common with null adenomas and other space-occupying lesions of the pituitary gland and surrounding region.
- Visual field abnormalities: Pressure on the optic chiasm due to a mass lesion produces bitemporal hemianopsia.
- Elevated intracranial pressure, which causes headache and nausea and vomiting.
- Obstructive hydrocephalus: Due to blockage of ventricular system.
- Cranial nerve palsies: Due to impingement upon adjacent cranial nerves.
- Diabetes insipidus: Due to destruction of posterior pituitary gland.
- Hypothalamic disturbances: Disorders of thirst, appetite, temperature regulation, behavior, and consciousness.
- Pituitary adenomas: Most often null cell adenomas are the type of adenoma that can grow to a size large enough to cause mass effect.
- Metastatic carcinoma.
- Craniopharyngioma
- Epidemiology: Two age ranges; one at 5–15 years of age and another at the sixth decade or older. Craniopharyngiomas are the most common cause of hypothalamic disturbances in children.
- Two types:Adamantinomatous (commonly calcify) and papillary (rarely calcify).
- Important points regarding craniopharyngioma
- Derived from Rathke pouch.
- Most are suprasellar.
- Prognosis is dependent upon completeness of surgical excision.
- Derived from Rathke pouch.
- Epidemiology: Two age ranges; one at 5–15 years of age and another at the sixth decade or older. Craniopharyngiomas are the most common cause of hypothalamic disturbances in children.
Pathology of the Posterior Pituitary Gland
Overview: The two syndromes most commonly associated with the posterior pituitary gland are syndrome of inappropriate antidiuretic hormone and diabetes insipidus, which are described below.
Basic description: Syndrome caused by hyperfunctioning of the posterior pituitary gland resulting in increased levels of antidiuretic hormone (ADH). Increased levels of ADH result in water retention and hyponatremia.
Causes of SIADH
- Ectopic ADH (commonly secreted by small cell lung carcinoma).
- Non-neoplastic diseases of the lung (e.g., tuberculosis, pneumonia).
- Central nervous system disorders (e.g., meningitis, abscess, head trauma).
- Injury to the hypothalamus or posterior pituitary gland.
Mechanism: Increased retention of water by increased ADH leads to hyponatremia.
Clinical presentation of SIADH
- Symptoms: Headache, anorexia, vomiting, confusion (when sodium level is 115–120 mEq/L), and stupor; coma and seizures (when sodium level is < 110 mEq/L). SIADH can cause delirium and dementia.
- Laboratory findings: Hyponatremia, decreased serum osmolarity, inappropriately concentrated urine (> 100 mOsm/kg), and low blood urea nitrogen (BUN).
Basic description: Syndrome caused by hypofunctioning of the posterior pituitary gland resulting in decreased levels of ADH.
Types of diabetes insipidus
- Central diabetes insipidus: Due to decreased production of ADH.
- Nephrogenic insipidus: Due to decreased renal responsiveness to ADH.
Causes of diabetes insipidus
- Central diabetes insipidus: Head trauma, neoplasms, Langerhans cell histiocytosis, infectious processes, surgical procedures, autoimmune diseases.
- Nephrogenic diabetes insipidus: Chronic renal disease; sickle cell anemia.
Clinical presentation of diabetes insipidus
- Symptoms: Polyuria and polydipsia.
- Laboratory testing: Dilute urine (specific gravity < 1.005); urine osmolality is < 200 mOsm/kg, and serum osmolality is increased. Administration of ADH will correct central diabetes insipidus, but not nephrogenic diabetes insipidus.
- Must differentiate fromprimary polydipsia (compulsive thirst). In diabetes insipidus, urine osmolarity is less than plasma osmolarity. In primary polydipsia, both are dilute. The definitive test is water deprivation, which causes decreased urine output and an increase in urine osmolarity in primary polydipsia and high urine output and dilute urine (decreased specific gravity) in diabetes insipidus.
Goiter
Overview: Enlargement of the thyroid gland, which can cause hyperthyroidism, hypothyroidism, or euthyroidism. The two types of goiter are endemic (occur in areas of iodine deficiency) and sporadic.
- Endemic goiter (i.e., goiters in more than 10% of population): The diet in the area is deficient in iodine. The thyroid gland enlarges so the individual can make the best use of limited iodine in the body. Patients may be euthyroid as a result, even though the gland is large.
- Sporadic goiter: Female predominance; young adults. Sporadic goiter is due to goitrogens such as brussel sprouts, cauliflower, and cabbage, which inhibit the formation of T3 and T4.
Pathogenesis of goiter: Low levels of thyroid hormones cause an increased level of TSH, which stimulates the thyroid gland, causing hyperplasia and hypertrophy of follicular cells. Alternating cycles of growth and degeneration lead to nodule formation.
Gross morphology of goiter: Enlarged thyroid gland with discrete nodules with a “glassy” texture, caused by the presence of abundant colloid (Figure 18-3).

Hyperthyroidism
Overview: Hyperthyroidism increases the basal metabolic rate and increases the organ’s sensitivity to catecholamines. Apathetic hyperthyroidism is most commonly seen in elderly patients, and is characterized by flat affect, weight loss, weakness, and emotional lability.
- Heat intolerance, sweating, and warm, flushed skin.
- Weight loss associated with increased appetite.
- Palpitations, tachycardia, tremor, anxiety, hyperactivity.
- Diarrhea.
- Fine hair.
- Important point: Tachycardia, tremor, and sweating are due to increased sensitivity to catecholamines
Feature | Hyperthyroidism | Hypothyroidism |
|---|---|---|
Temperature insensitivity | Heat intolerant | Cold intolerant |
Weight | Lose weight, but have increased appetite | Gain weight, despite decreased appetite |
Activity | Anxious, hyperactive | Depression, sluggish |
Gastrointestinal function | Diarrhea | Constipation |
Quality of hair | Fine | Coarse |
Laboratory findings of primary hyperthyroidism: Decreased level of TSH and elevated T4; occasionally, patients will have only an elevated T3.
Causes of primary hyperthyroidism: The two more common causes of primary hyperthyroidism, Graves disease and toxic goiter, will be discussed below. A list of other causes of primary hyperthyroidism will follow that discussion.
Pathogenesis: Due to anti–TSH receptor antibody that stimulates the receptor, resulting in the production of T3 and T4.
Epidemiology: Occurs between 20 and 40 years of age; predominance of female to male, with a ratio of 7:1.
HLA associations: HLA-B8 and HLA-DR3.
Morphology of Graves disease
- Gross: Thyroid gland itself is diffusely enlarged but not nodular.
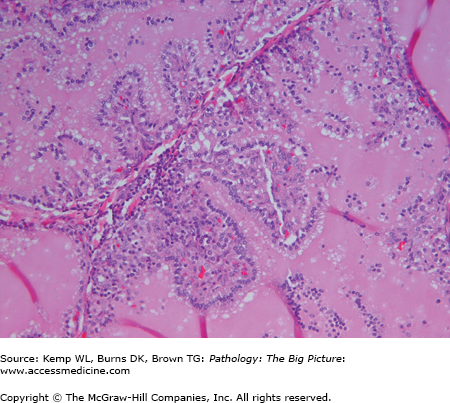
- Microscopic: Hyperplastic follicles with papillae (Figure 18-4). The papillae do not have fibrovascular cores. “Scalloping” of colloid at the periphery of the follicles.
Clinical presentation of Graves disease
- Signs and symptoms of hyperthyroidism.
- Exophthalmos: Protrusion of eye due to retro-orbital soft tissue infiltrated with lymphocytes, edema, and increased amount of glycosaminoglycans. May progress even after hyperthyroidism is under control.
- Pretibial myxedema: Scaling and thickening of dermis by deposition of glycosaminoglycans and lymphocytes. No pitting occurs.
- Important point: Exophthalmos and pretibial myxedema are specific to Graves disease and are not found in other causes of hyperthyroidism.
Toxic (diffuse or multinodular) goiter: A goiter that usually has one or more nodules that produce T3 and T4. However, most goiters are not hyperfunctioning.
Rare causes of primary hyperthyroidism: Thyroid adenoma, postpartum thyroiditis, amiodarone toxicity, and iodinated contrast media.
Causes of secondary hyperthyroidism:Struma ovarii (i.e., ovarian teratoma composed of thyroid tissue) and hydatidiform mole (i.e., proliferation of trophoblasts that cause excessive production of chorionic gonadotropin, which has intrinsic TSH-like activity).
Basic description: Abrupt onset of severe hyperthyroidism.
Cause of thyroid storm: Most commonly associated with Graves disease in patients with secondarily increased levels of catecholamines due to surgery and acute infection.
Complications of thyroid storm: Can cause cardiac dysrhythmias and sudden death.
Clinical presentation of thyroid storm: Fever, flushing, and sweating. Patients can have agitation, marked weakness, and mental status changes. Hyperthermia out of proportion to other findings is characteristic of thyroid storm. Patients can also have tachycardia, atrial fibrillation, and delirium.
Hypothyroidism
- Weight gain.
- Cold intolerance and cool skin.
- Thinning hair and loss of lateral portion of eyebrows (known as the “Queen Anne sign

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


