OBJECTIVES
After reading this chapter, you should be able to:
List the hormones that affect the plasma glucose concentration and briefly describe the action of each.
Describe the structure of the pancreatic islets and name the hormones secreted by each of the cell types in the islets.
Describe the structure of insulin and outline the steps involved in its biosynthesis and release into the bloodstream.
List the consequences of insulin deficiency and explain how each of these abnormalities is produced.
Describe insulin receptors, the way they mediate the effects of insulin, and the way they are regulated.
Describe the types of glucose transporters found in the body and the function of each.
List the major factors that affect the secretion of insulin.
Describe the structure of glucagon and other physiologically active peptides produced from its precursor.
List the physiologically significant effects of glucagon and the factors that regulate glucagon secretion.
Describe the physiologic effects of somatostatin in the pancreas.
Outline the mechanisms by which thyroid hormones, adrenal glucocorticoids, catecholamines, and growth hormone affect carbohydrate metabolism.
Understand the major differences between type 1 and type 2 diabetes.
INTRODUCTION
At least four polypeptides with regulatory activity are secreted by the islets of Langerhans in the pancreas. Two of these, insulin and glucagon, are hormones and have important functions in the regulation of the intermediary metabolism of carbohydrates, proteins, and fats. The third polypeptide, somatostatin, plays a role in the regulation of islet cell secretion, and the fourth, pancreatic polypeptide, is probably concerned primarily with the regulation of ion transport in the intestine. Glucagon, somatostatin, and possibly pancreatic polypeptide are also secreted by cells in the mucosa of the gastrointestinal tract.
Insulin is anabolic, increasing the storage of glucose, fatty acids, and amino acids. Glucagon is catabolic, mobilizing glucose, fatty acids, and the amino acids from stores into the bloodstream. The two hormones are thus reciprocal in their overall action and are reciprocally secreted in most circumstances. Insulin excess causes hypoglycemia, which leads to convulsions and coma. Insulin deficiency, either absolute or relative, causes diabetes mellitus (chronic elevated blood glucose), a complex and debilitating disease that if untreated is eventually fatal. Glucagon deficiency can cause hypoglycemia, and glucagon excess makes diabetes worse. Excess pancreatic production of somatostatin causes hyperglycemia and other manifestations of diabetes.
A variety of other hormones also have important roles in the regulation of carbohydrate metabolism.
ISLET CELL STRUCTURE
The islets of Langerhans (Figure 24–1) are ovoid, 76- × 175-μm collections of cells. The islets are scattered throughout the pancreas, although they are more plentiful in the tail than in the body and head. β-Islets make up about 2% of the volume of the gland, whereas the exocrine portion of the pancreas (see Chapter 25) makes up 80%, and ducts and blood vessels make up the remainder. Humans have 1 to 2 million islets. Each has a copious blood supply; blood from the islets, like that from the gastrointestinal tract (but unlike that from any other endocrine organs) drains into the hepatic portal vein.
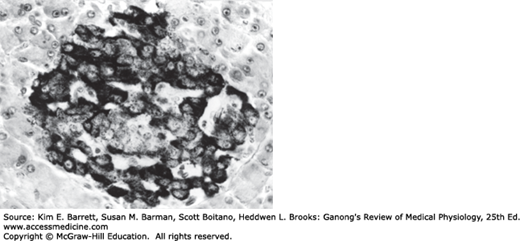
The cells in the islets can be divided into types on the basis of their staining properties and morphology. Humans have at least four distinct cell types: A, B, D, and F cells. A, B, and D cells are also called α, β, and δ cells. However, this leads to confusion in view of the use of Greek letters to refer to other structures in the body, particularly adrenergic receptors (see Chapter 7). The A cells secrete glucagon, the B cells secrete insulin, the D cells secrete somatostatin, and the F cells secrete pancreatic polypeptide. The B cells, which are the most common and account for 60–75% of the cells in the islets, are generally located in the center of each islet. They tend to be surrounded by the A cells, which make up 20% of the total, and the less common D and F cells. The islets in the tail, the body, and the anterior and superior part of the head of the human pancreas have many A cells and few if any F cells in the outer rim, whereas in rats and probably in humans, the islets in the posterior part of the head of the pancreas have a relatively large number of F cells and few A cells. The A-cell-rich (glucagon-rich) islets arise embryologically from the dorsal pancreatic bud, and the F-cell-rich (pancreatic polypeptide-rich) islets arise from the ventral pancreatic bud. These buds arise separately from the duodenum.
The B cell granules are packets of insulin in the cell cytoplasm. The shape of the packets varies from species to species; in humans, some are round whereas others are rectangular (Figure 24–2). In the B cells, the insulin molecule forms polymers and also complexes with zinc. The differences in the shape of the packets are probably due to differences in the size of polymers or zinc aggregates of insulin. The A granules, which contain glucagon, are relatively uniform from species to species (Figure 24–3). The D cells also contain large numbers of relatively homogeneous granules.
FIGURE 24–2
Electronmicrograph of two adjoining B cells in a human pancreatic islet. The B granules are the crystals in the membrane-lined vesicles. They vary in shape from rhombic to round (× 26,000). (Reproduced with permission from Fawcett DW: Bloom and Fawcett A Textbook of Histology, 11th ed. St. Louis, MO: Saunders; 1986.)
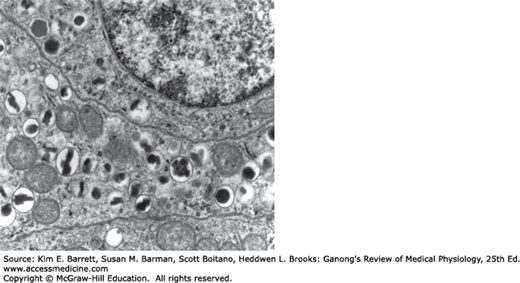
FIGURE 24–3
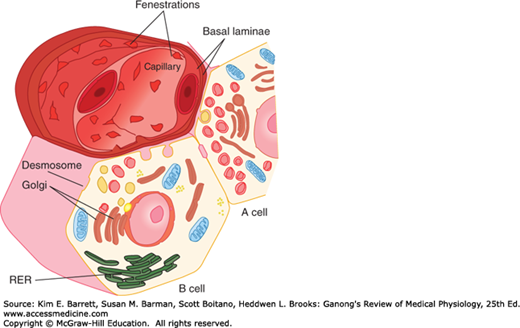
A and B cells, showing their relation to a blood vessel. Insulin from the B cell and glucagon from the A cell are secreted by exocytosis and cross the basal lamina of the cell and the basal lamina of the capillary before entering the lumen of the fenestrated capillary. RER, rough endoplasmic reticulum. (Reproduced with permission from Junqueira IC, Carneiro J: Basic Histology: Text and Atlas, 10th ed. New York, NY: McGraw-Hill; 2003.)
STRUCTURE, BIOSYNTHESIS, & SECRETION OF INSULIN
Insulin is a polypeptide containing two chains of amino acids linked by disulfide bridges. Minor differences occur in the amino acid composition of the molecule from species to species. The differences are generally not sufficient to affect the biologic activity of a particular insulin in heterologous species but are sufficient to make the insulin antigenic. If insulin of one species is injected for a prolonged period into another species, the anti-insulin antibodies formed inhibit the injected insulin. Almost all humans who have received commercial bovine insulin for more than 2 months have antibodies against bovine insulin, but the titer is usually low. Porcine insulin differs from human insulin by only one amino acid residue and has low antigenicity. Human insulin produced in bacteria by recombinant DNA technology is now widely used to avoid antibody formation.
Insulin is synthesized in the rough endoplasmic reticulum of the B cells (Figure 24–3). It is then transported to the Golgi apparatus, where it is packaged into membrane-bound granules. These granules move to the plasma membrane by a process involving microtubules, and their contents are expelled by exocytosis (see Chapters 2 and 16). The insulin then crosses the basal lamina of the B cell and a neighboring capillary and the fenestrated endothelium of the capillary to reach the bloodstream. The fenestrations are discussed in detail in Chapter 31.
Like other polypeptide hormones and related proteins that enter the endoplasmic reticulum, insulin is synthesized as part of a larger preprohormone (see Chapter 1). The gene for insulin is located on the short arm of chromosome 11 in humans. It has two introns and three exons. Preproinsulin originates from the endoplasmic reticulum. The remainder of the molecule is then folded, and the disulfide bonds are formed to make proinsulin. The peptide segment connecting the A and B chains, the connecting peptide (C peptide), facilitates the folding and then is detached in the granules before secretion. Two proteases are involved in processing the proinsulin. Normally, 90–97% of the product released from the B cells is insulin along with equimolar amounts of C peptide. The rest is mostly proinsulin. C peptide can be measured by radioimmunoassay, and its level in blood provides an index of B cell function in patients receiving exogenous insulin.
FATE OF SECRETED INSULIN
Plasma contains a number of substances with insulin-like activity in addition to insulin. The activity that is not suppressed by anti-insulin antibodies has been called nonsuppressible insulin-like activity (NSILA). Most, if not all, of this activity persists after pancreatectomy and is due to the insulin-like growth factors IGF-I and IGF-II (see Chapter 18). These IGFs are polypeptides. Small amounts are free in the plasma (low-molecular-weight fraction), but large amounts are bound to proteins (high-molecular-weight fraction).
One may well ask why pancreatectomy causes diabetes mellitus when NSILA persists in the plasma. However, the insulin-like activities of IGF-I and IGF-II are weak compared to that of insulin and likely subserve other specific functions.
The half-life of insulin in the circulation in humans is about 5 min. Insulin binds to insulin receptors, and some is internalized. It is destroyed by proteases in the endosomes formed by the endocytotic process.
The physiologic effects of insulin are far-reaching and complex. They are conveniently divided into rapid, intermediate, and delayed actions (Table 24–1). The best known is the hypoglycemic effect, but there are additional effects on amino acid and electrolyte transport, many enzymes, and growth. The net effect of the hormone is storage of carbohydrate, protein, and fat. Therefore, insulin is appropriately called the “hormone of abundance.”
Rapid (seconds)
|
Intermediate (minutes)
|
Delayed (hours)
|
The actions of insulin on adipose tissue; skeletal, cardiac, and smooth muscle; and the liver are summarized in Table 24–2.
Adipose tissue
|
Muscle
|
Liver
|
General
|
Glucose enters cells by facilitated diffusion (see Chapter 1) or, in the intestine and kidneys, by secondary active transport with Na+. In muscle, adipose, and some other tissues, insulin stimulates glucose entry into cells by increasing the number of glucose transporters (GLUTs) in the cell membranes.
The GLUTs that are responsible for facilitated diffusion of glucose across cell membranes are a family of closely related proteins that span the cell membrane 12 times and have their amino and carboxyl terminals inside the cell. They differ from and have no homology with the sodium-glucose linked transporters (SGLT-1 and SGLT-2), which are responsible for the secondary active transport of glucose in the intestine (see Chapter 26) and renal tubules (see Chapter 38), although the SGLTs also have 12 transmembrane domains.
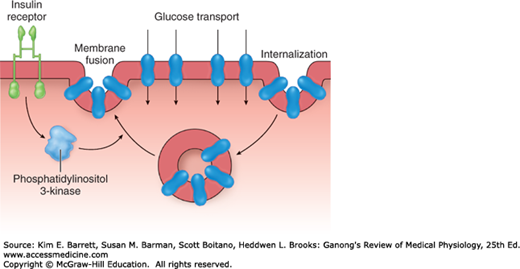
Seven different GLUTs, named GLUT 1–7 in order of discovery, have been characterized (Table 24–3). They contain 492–524 amino acid residues and their affinity for glucose varies. Each transporter appears to have evolved for special tasks. GLUT-4 is the transporter in muscle and adipose tissue that is stimulated by insulin. A pool of GLUT-4 molecules is maintained within vesicles in the cytoplasm of insulin-sensitive cells. When the insulin receptors of these cells are activated, the vesicles move rapidly to the cell membrane and fuse with it, inserting the transporters into the cell membrane (Figure 24–4). When insulin action ceases, the transporter-containing patches of membrane are endocytosed and the vesicles are ready for the next exposure to insulin. Activation of the insulin receptor brings about the movement of the vesicles to the cell membrane by activating phosphatidylinositol 3-kinase (Figure 24–4). Most of the other GLUT transporters that are not insulin-sensitive appear to be constitutively expressed in the cell membrane.
| Function | Km(mM)a | Major Sites of Expression | |
|---|---|---|---|
| Secondary active transport (Na1–glucose cotransport) | |||
| SGLT 1 | Absorption of glucose | 0.1–1.0 | Small intestine, renal tubules |
| SGLT 2 | Absorption of glucose | 1.6 | Renal tubules |
| Facilitated diffusion | |||
| GLUT 1 | Basal glucose uptake | 1–2 | Placenta, blood-brain barrier, brain, red cells, kidneys, colon, many other organs |
| GLUT 2 | B-cell glucose sensor; transport outofintestinal and renal epithelialcells | 12–20 | B cells of islets, liver, epithelial cells of small intestine, kidneys |
| GLUT 3 | Basal glucose uptake | <1 | Brain, placenta, kidneys, many other organs |
| GLUT 4 | Insulin-stimulated glucose uptake | 5 | Skeletal and cardiac muscle, adipose tissue, other tissues |
| GLUT 5 | Fructose transport | 1–2 | Jejunum, sperm |
| GLUT 6 | Unknown | — | Brain, spleen and leukocytes |
| GLUT 7 | Glucose 6-phosphate transporter in endoplasmic reticulum | — | Liver |
FIGURE 24–4
Cycling of GLUT-4 transporters through endosomes in insulin-sensitive tissues. Activation of the insulin receptor causes activation of phosphatidylinositol 3-kinase, which speeds translocation of the GLUT-4–containing endosomes into the cell membrane. The GLUT-4 transporters then mediate glucose transport into the cell.
In the tissues in which insulin increases the number of GLUTs in cell membranes, the rate of phosphorylation of the glucose, once it has entered the cells, is regulated by other hormones. Growth hormone and cortisol both inhibit phosphorylation in certain tissues. Transport is normally so rapid that it is not a rate-limiting step in glucose metabolism. However, it is rate-limiting in B cells.
Insulin also increases the entry of glucose into liver cells, but it does not exert this effect by increasing the number of GLUT-4 transporters in the cell membranes. Instead, it induces glucokinase, and this increases the phosphorylation of glucose, so that the intracellular free glucose concentration stays low, facilitating the entry of glucose into the cell.
Insulin-sensitive tissues also contain a population of GLUT-4 vesicles that move into the cell membrane in response to exercise, a process that occurs independent of the action of insulin. This is why exercise lowers blood sugar. A 5’-adenosine monophosphate (AMP)–activated kinase may trigger the insertion of these vesicles into the cell membrane.
The maximal decline in plasma glucose occurs 30 min after intravenous injection of insulin. After subcutaneous administration, the maximal fall occurs in 2–3 h. A wide variety of insulin preparations are now available commercially. These include insulins that have been complexed with protamine and other polypeptides to delay absorption and degradation, and synthetic insulins in which there have been changes in amino acid residues. In general, they fall into three categories: rapid, intermediate-acting, and long-acting (24–36 h).
Insulin causes K+ to enter cells, with a resultant lowering of the extracellular K+ concentration. Infusions of insulin and glucose significantly lower the plasma K+ level in normal individuals and are very effective for the temporary relief of hyperkalemia in patients with renal failure. Hypokalemia often develops when patients with diabetic acidosis are treated with insulin. The reason for the intracellular migration of K+ is still uncertain. However, insulin increases the activity of Na, K ATPase in cell membranes, so that more K+ is pumped into cells.
The hypoglycemic and other effects of insulin are summarized in temporal terms in Table 24–1, and the net effects on various tissues are summarized in Table 24–2. The action on glycogen synthase fosters glycogen storage, and the actions on glycolytic enzymes favor glucose metabolism to two carbon fragments (see Chapter 1), with resulting promotion of lipogenesis. Stimulation of protein synthesis from amino acids entering the cells and inhibition of protein degradation foster growth.
The anabolic effect of insulin is aided by the protein-sparing action of adequate intracellular glucose supplies. Failure to grow is a symptom of diabetes in children, and insulin stimulates the growth of immature hypophysectomized rats to almost the same degree as growth hormone.
MECHANISM OF ACTION
Insulin receptors are found on many different cells in the body, including cells in which insulin does not increase glucose uptake.
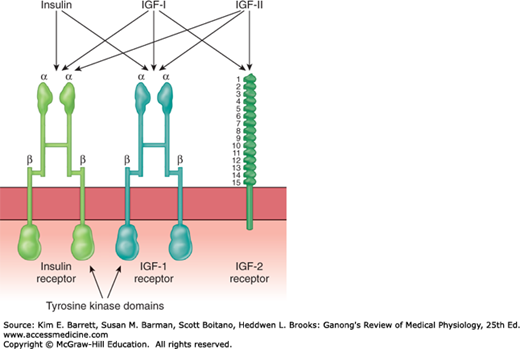
The insulin receptor, which has a molecular weight of approximately 340,000, is a tetramer made up of two α and two β glycoprotein subunits (Figure 24–5). All these are synthesized on a single mRNA and then proteolytically separated and bound to each other by disulfide bonds. The gene for the insulin receptor has 22 exons and in humans is located on chromosome 19. The α subunits bind insulin and are extracellular, whereas the β subunits span the membrane. The intracellular portions of the β subunits have tyrosine kinase activity. The α and β subunits are both glycosylated, with sugar residues extending into the interstitial fluid.
FIGURE 24–5
Insulin, IGF-I, and IGF-II receptors. Each hormone binds primarily to its own receptor, but insulin also binds to the IGF-I receptor, and IGF-I and IGF-II bind to all three. The purple boxes are intracellular tyrosine kinase domains. Note the marked similarity between the insulin receptor and the IGF-I receptor; also note the 15 repeat sequences in the extracellular portion of the IGF-II receptor. ISF, interstitial fluid.
Binding of insulin triggers the tyrosine kinase activity of the β subunits, producing autophosphorylation of the β subunits on tyrosine residues. The autophosphorylation, which is necessary for insulin to exert its biologic effects, triggers phosphorylation of some cytoplasmic proteins and dephosphorylation of others, mostly on serine and threonine residues. Insulin receptor substrate (IRS-1) mediates some of the effects in humans but there are other effector systems as well (Figure 24–6). For example, mice in which the insulin receptor gene is knocked out show marked growth retardation in utero, have abnormalities of the central nervous system (CNS) and skin, and die at birth of respiratory failure, whereas IRS-1 knockouts show only moderate growth retardation in utero, survive, and are insulin-resistant but otherwise nearly normal.
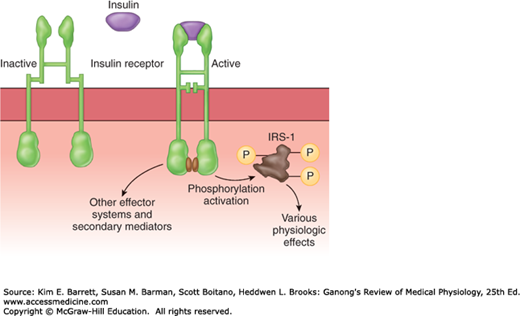
The growth-promoting protein anabolic effects of insulin are mediated via phosphatidylinositol 3-kinase (PI3K), and evidence indicates that in invertebrates, this pathway is involved in the growth of nerve cells and axon guidance in the visual system.
It is interesting to compare the insulin receptor with other related receptors. The insulin receptor is very similar to the receptor for IGF-I but different from the receptor for IGF-II (Figure 24–5). Other receptors for growth factors and receptors for various oncogenes also are tyrosine kinases. However, the amino acid composition of these receptors is quite different.
When insulin binds to its receptors, they aggregate in patches and are taken up into the cell by receptor-mediated endocytosis (see Chapter 2). Eventually, the insulin–receptor complexes enter lysosomes, where the receptors are broken down or recycled. The half-life of the insulin receptor is about 7 h.
CONSEQUENCES OF INSULIN DEFICIENCY
The far-reaching physiologic effects of insulin are highlighted by a consideration of the extensive and serious consequences of insulin deficiency (Clinical Box 24–1).
In humans, insulin deficiency is a common pathologic condition. In animals, it can be produced by pancreatectomy; by administration of alloxan, streptozocin, or other toxins that in appropriate doses cause selective destruction of the B cells of the pancreatic islets; by administration of drugs that inhibit insulin secretion; and by administration of anti-insulin antibodies. Strains of mice, rats, hamsters, guinea pigs, miniature swine, and monkeys that have a high incidence of spontaneous diabetes mellitus have also been described.
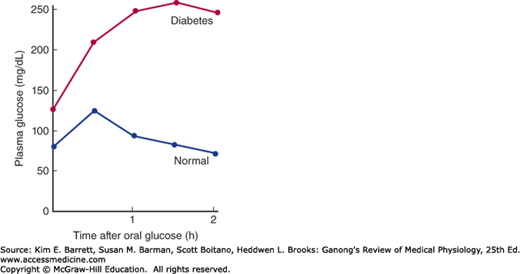
In diabetes, glucose piles up in the bloodstream, especially after meals. If a glucose load is given to a diabetic, the plasma glucose rises higher and returns to the baseline more slowly than it does in normal individuals. The response to a standard oral test dose of glucose, the oral glucose tolerance test, is used in the clinical diagnosis of diabetes (Figure 24–7).
FIGURE 24–7
Oral glucose tolerance test. Adults are given 75 g of glucose in 300 mL of water. In normal individuals, the fasting venous plasma glucose is less than 115 mg/dL, the 2-hour value is less than 140 mg/dL, and no value is greater than 200 mg/dL. Diabetes mellitus is present if the 2-hour value and one other value are greater than 200 mg/dL. Impaired glucose tolerance is diagnosed when the values are above the upper limits of normal but below the values diagnostic of diabetes.
Impaired glucose tolerance in diabetes is due in part to reduced entry of glucose into cells (decreased peripheral utilization). In the absence of insulin, the entry of glucose into skeletal, cardiac, and smooth muscle and other tissues is decreased (Figure 24–8). Glucose uptake by the liver is also reduced, but the effect is indirect. Intestinal absorption of glucose is unaffected, as is its reabsorption from the urine by the cells of the proximal tubules of the kidneys. Glucose uptake by most of the brain and the red blood cells is also normal.
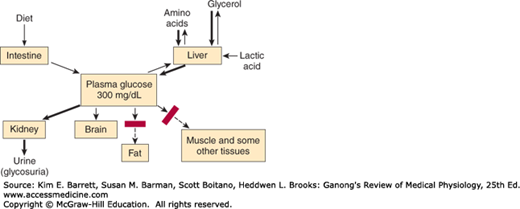
The second and the major cause of hyperglycemia in diabetes is derangement of the glucostatic function of the liver (see Chapter 28). The liver takes up glucose from the bloodstream and stores it as glycogen, but because the liver contains glucose-6-phosphatase it also discharges glucose into the bloodstream. Insulin facilitates glycogen synthesis and inhibits hepatic glucose output. When the plasma glucose is high, insulin secretion is normally increased and hepatic glucogenesis is decreased. This response does not occur in type 1 diabetes mellitus (as insulin is absent) and in type 2 diabetes mellitus (as tissues are insulin-resistant). Glucagon can contribute to hyperglycemia as it stimulates gluconeogenesis. Glucose output by the liver can be stimulated by catecholamines, cortisol, and growth hormone (ie, during a stress response).
CLINICAL BOX 24–1 Diabetes Mellitus
The constellation of abnormalities caused by insulin deficiency is called diabetes mellitus. Greek and Roman physicians used the term “diabetes” to refer to conditions in which the cardinal finding was a large urine volume, and two types were distinguished: “diabetes mellitus,” in which the urine tasted sweet; and “diabetes insipidus,” in which the urine had little taste. Today, the term “diabetes insipidus” is reserved for conditions in which there is a deficiency of the production or action of vasopressin (see Chapter 38), and the unmodified word “diabetes” is generally used as a synonym for diabetes mellitus.
The cause of clinical diabetes is always a deficiency of the effects of insulin at the tissue level. Type 1 diabetes, or insulin-dependent diabetes mellitus (IDDM), is due to insulin deficiency caused by autoimmune destruction of the B cells in the pancreatic islets, and it accounts for 3–5% of cases and usually presents in children. Type 2 diabetes, or non–insulin-dependent diabetes mellitus (NIDDM), is characterized by the dysregulation of insulin release from the B cells, along with insulin resistance in peripheral tissues such as skeletal muscle, brain, and liver. Type 2 diabetes historically presented in overweight or obese adults, although it is increasingly being diagnosed in children as childhood obesity increases.
Diabetes is characterized by polyuria (passage of large volumes of urine), polydipsia (excessive drinking), weight loss in spite of polyphagia (increased appetite), hyperglycemia, glycosuria, ketosis, acidosis, and coma. Widespread biochemical abnormalities are present, but the fundamental defects to which most of the abnormalities can be traced are (1) reduced entry of glucose into various “peripheral” tissues and (2) increased liberation of glucose into the circulation from the liver. Therefore, there is an extracellular glucose excess and, in many cells, an intracellular glucose deficiency—a situation that has been called “starvation in the midst of plenty.” Also, the entry of amino acids into muscle is decreased and lipolysis is increased.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


