HIGH-ANION GAP ACIDOSES
Lactic Acidosis An increase in plasma l-lactate may be secondary to poor tissue perfusion (type A)—circulatory insufficiency (shock, cardiac failure), severe anemia, mitochondrial enzyme defects, and inhibitors (carbon monoxide, cyanide)—or to aerobic disorders (type B)—malignancies, nucleoside analogue reverse transcriptase inhibitors in HIV, diabetes mellitus, renal or hepatic failure, thiamine deficiency, severe infections (cholera, malaria), seizures, or drugs/toxins (biguanides, ethanol, methanol, propylene glycol, isoniazid, and fructose). Unrecognized bowel ischemia or infarction in a patient with severe atherosclerosis or cardiac decompensation receiving vasopressors is a common cause of lactic acidosis. Pyroglutamic acidemia has been reported in critically ill patients receiving acetaminophen, which is associated with depletion of glutathione. D-Lactic acid acidosis, which may be associated with jejunoileal bypass, short bowel syndrome, or intestinal obstruction, is due to formation of D-lactate by gut bacteria.
Ketoacidosis • DIABETIC KETOACIDOSIS (DKA) This condition is caused by increased fatty acid metabolism and the accumulation of ketoacids (acetoacetate and β-hydroxybutyrate). DKA usually occurs in insulin-dependent diabetes mellitus in association with cessation of insulin or an intercurrent illness such as an infection, gastroenteritis, pancreatitis, or myocardial infarction, which increases insulin requirements temporarily and acutely. The accumulation of ketoacids accounts for the increment in the AG and is accompanied most often by hyperglycemia (glucose >17 mmol/L [300 mg/dL]). The relationship between the ΔAG and ΔHCO3– is typically ~1:1 in DKA. It should be noted that, because insulin prevents production of ketones, bicarbonate therapy is rarely needed except with extreme acidemia (pH < 7.1), and then in only limited amounts. Patients with DKA are typically volume depleted and require fluid resuscitation with isotonic saline. Volume overexpansion with IV fluid administration is not uncommon, however, and contributes to the development of a hyperchloremic acidosis during treatment of DKA. The mainstay for treatment of this condition is IV regular insulin and is described in Chap. 417 in more detail.
ALCOHOLIC KETOACIDOSIS (AKA) Chronic alcoholics can develop ketoacidosis when alcohol consumption is abruptly curtailed and nutrition is poor. AKA is usually associated with binge drinking, vomiting, abdominal pain, starvation, and volume depletion. The glucose concentration is variable, and acidosis may be severe because of elevated ketones, predominantly β-hydroxybutyrate. Hypoperfusion may enhance lactic acid production, chronic respiratory alkalosis may accompany liver disease, and metabolic alkalosis can result from vomiting (refer to the relationship between ΔAG and ΔHCO3–). Thus, mixed acid-base disorders are common in AKA. As the circulation is restored by administration of isotonic saline, the preferential accumulation of β-hydroxybutyrate is then shifted to acetoacetate. This explains the common clinical observation of an increasingly positive nitroprusside reaction as the patient improves. The nitroprusside ketone reaction (Acetest) can detect acetoacetic acid but not β-hydroxybutyrate, so that the degree of ketosis and ketonuria can not only change with therapy, but can be underestimated initially. Patients with AKA usually present with relatively normal renal function, as opposed to DKA, where renal function is often compromised because of volume depletion (osmotic diuresis) or diabetic nephropathy. The AKA patient with normal renal function may excrete relatively large quantities of ketoacids in the urine and, therefore, may have a relatively normal AG and a discrepancy in the ΔAG/ΔHCO3– relationship.
Drug- and Toxin-Induced Acidosis • SALICYLATES (See also Chap. 472e) Salicylate intoxication in adults usually causes respiratory alkalosis or a mixture of high-AG metabolic acidosis and respiratory alkalosis. Only a portion of the AG is due to salicylates. Lactic acid production is also often increased.
ALCOHOLS Under most physiologic conditions, sodium, urea, and glucose generate the osmotic pressure of blood. Plasma osmolality is calculated according to the following expression: Posm = 2Na+ + Glu + BUN (all in mmol/L), or, using conventional laboratory values in which glucose and BUN are expressed in milligrams per deciliter: Posm = 2Na+ + Glu/18 + BUN/2.8. The calculated and determined osmolality should agree within 10–15 mmol/kg H2O. When the measured osmolality exceeds the calculated osmolality by >10–15 mmol/kg H2O, one of two circumstances prevails. Either the serum sodium is spuriously low, as with hyperlipidemia or hyperproteinemia (pseudohyponatremia), or osmolytes other than sodium salts, glucose, or urea have accumulated in plasma. Examples of such osmolytes include mannitol, radiocontrast media, ethanol, isopropyl alcohol, ethylene glycol, propylene glycol, methanol, and acetone. In this situation, the difference between the calculated osmolality and the measured osmolality (osmolar gap) is proportional to the concentration of the unmeasured solute. With an appropriate clinical history and index of suspicion, identification of an osmolar gap is helpful in identifying the presence of poison-associated AG acidosis. Three alcohols may cause fatal intoxications: ethylene glycol, methanol, and isopropyl alcohol. All cause an elevated osmolal gap, but only the first two cause a high-AG acidosis.
ETHYLENE GLYCOL (See also Chap. 472e) Ingestion of ethylene glycol (commonly used in antifreeze) leads to a metabolic acidosis and severe damage to the CNS, heart, lungs, and kidneys. The increased AG and osmolar gap are attributable to ethylene glycol and its metabolites, oxalic acid, glycolic acid, and other organic acids. Lactic acid production increases secondary to inhibition of the tricarboxylic acid cycle and altered intracellular redox state. Diagnosis is facilitated by recognizing oxalate crystals in the urine, the presence of an osmolar gap in serum, and a high-AG acidosis. Although use of a Wood’s lamp to visualize the fluorescent additive to commercial antifreeze in the urine of patients with ethylene glycol ingestion, this is rarely reproducible. The combination of a high AG and high osmolar gap in a patient suspected of ethylene glycol ingestion should be taken as evidence of ethylene glycol toxicity. Treatment should not be delayed while awaiting measurement of ethylene glycol levels in this setting.
METHANOL (See also Chap. 472e) The ingestion of methanol (wood alcohol) causes metabolic acidosis, and its metabolites formaldehyde and formic acid cause severe optic nerve and CNS damage. Lactic acid, ketoacids, and other unidentified organic acids may contribute to the acidosis. Due to its low molecular mass (32 Da), an osmolar gap is usually present.
PROPYLENE GLYCOL Propylene glycol is the vehicle used in IV administration of diazepam, lorazepam, phenobarbital, nitroglycerine, etomidate, enoximone, and phenytoin. Propylene glycol is generally safe for limited use in these IV preparations, but toxicity has been reported, most often in the setting of the intensive care unit in patients receiving frequent or continuous therapy. This form of high-gap acidosis should be considered in patients with unexplained high-gap acidosis, hyperosmolality, and clinical deterioration. Propylene glycol, like ethylene glycol and methanol, is metabolized by alcohol dehydrogenase. With intoxication by propylene glycol, the first response is to stop the offending infusion. Additionally, fomepizole should also be administered in acidotic patients.
ISOPROPYL ALCOHOL Ingested isopropanol is absorbed rapidly and may be fatal when as little as 150 mL of rubbing alcohol, solvent, or deicer is consumed. A plasma level >400 mg/dL is life-threatening. Isopropyl alcohol is metabolized by alcohol dehydrogenase to acetone. The characteristic features differ from ethylene glycol and methanol in that the parent compound, not the metabolites, causes toxicity, and an AG acidosis is not present because acetone is rapidly excreted. Both isopropyl alcohol and acetone increase the osmolal gap, and hypoglycemia is common. Alternative diagnoses should be considered if the patient does not improve significantly within a few hours. Patients with hemodynamic instability with plasma levels above 400 mg/dL should be considered for hemodialysis.
PYROGLUTAMIC ACID Acetaminophen-induced high-AG metabolic acidosis is uncommon but is being recognized more often in either patients with acetaminophen overdose or malnourished or critically ill patients receiving acetaminophen in typical dosage. 5-Oxoproline accumulation after acetaminophen should be suspected in the setting of an unexplained high-AG acidosis without elevation of the osmolar gap in patients receiving acetaminophen. The first step in treatment is to immediately discontinue the drug. Additionally, sodium bicarbonate IV should be given. Although N-acetylcysteine has been suggested, it is not known if it hastens the metabolism of 5-oxoproline by increasing intracellular glutathione concentrations in this setting.
Renal Failure (See also Chap. 335) The hyperchloremic acidosis of moderate renal insufficiency is eventually converted to the high-AG acidosis of advanced renal failure. Poor filtration and reabsorption of organic anions contribute to the pathogenesis. As renal disease progresses, the number of functioning nephrons eventually becomes insufficient to keep pace with net acid production. Uremic acidosis is characterized, therefore, by a reduced rate of NH4+ production and excretion. The acid retained in chronic renal disease is buffered by alkaline salts from bone. Despite significant retention of acid (up to 20 mmol/d), the serum [HCO3–] does not decrease further, indicating participation of buffers outside the extracellular compartment. Chronic metabolic acidosis results in significant loss of bone mass due to reduction in bone calcium carbonate. Chronic acidosis also increases urinary calcium excretion, proportional to cumulative acid retention.
NON–ANION GAP METABOLIC ACIDOSES
Alkali can be lost from the gastrointestinal tract from diarrhea or from the kidneys (renal tubular acidosis, RTA). In these disorders (Table 66-5), reciprocal changes in [Cl–] and [HCO3–] result in a normal AG. In pure non–AG acidosis, therefore, the increase in [Cl–] above the normal value approximates the decrease in [HCO3–]. The absence of such a relationship suggests a mixed disturbance.
CAUSES OF NON–ANION GAP ACIDOSIS |
Abbreviations: ACE-I, angiotensin-converting enzyme inhibitor; ARB, angiotensin receptor blocker; PHA, pseudohypoaldosteronism; RTA, renal tubular acidosis.
Proximal RTA (type 2 RTA) (Chap. 339) is most often due to generalized proximal tubular dysfunction manifested by glycosuria, generalized aminoaciduria, and phosphaturia (Fanconi syndrome). With a low plasma [HCO3–], the urine pH is acid (pH <5.5). The fractional excretion of [HCO3–] may exceed 10–15% when the serum HCO3– >20 mmol/L. Because HCO3– is not reabsorbed normally in the proximal tubule, therapy with NaHCO3 will enhance renal potassium wasting and hypokalemia.
The typical findings in acquired or inherited forms of classic distal RTA (type 1 RTA) include hypokalemia, non-AG metabolic acidosis, low urinary NH4+ excretion (positive UAG, low urine [NH4+]), and inappropriately high urine pH (pH > 5.5). Most patients have hypocitraturia and hypercalciuria, so nephrolithiasis, nephrocalcinosis, and bone disease are common. In generalized distal RTA (type 4 RTA), hyperkalemia is disproportionate to the reduction in glomerular filtration rate (GFR) because of coexisting dysfunction of potassium and acid secretion. Urinary ammonium excretion is invariably depressed, and renal function may be compromised, for example, due to diabetic nephropathy, obstructive uropathy, or chronic tubulointerstitial disease.
Hyporeninemic hypoaldosteronism typically causes non-AG metabolic acidosis, most commonly in older adults with diabetes mellitus or tubulointerstitial disease and renal insufficiency. Patients usually have mild to moderate CKD (GFR, 20–50 mL/min) and acidosis, with elevation in serum [K+] (5.2–6.0 mmol/L), concurrent hypertension, and congestive heart failure. Both the metabolic acidosis and the hyperkalemia are out of proportion to impairment in GFR. Nonsteroidal anti-inflammatory drugs, trimethoprim, pentamidine, and angiotensin-converting enzyme (ACE) inhibitors can also cause non-AG metabolic acidosis in patients with renal insufficiency (Table 66-5).
METABOLIC ALKALOSIS
Metabolic alkalosis is manifested by an elevated arterial pH, an increase in the serum [HCO3–], and an increase in PaCO2 as a result of compensatory alveolar hypoventilation (Table 66-1). It is often accompanied by hypochloremia and hypokalemia. The arterial pH establishes the diagnosis, because it is increased in metabolic alkalosis and decreased or normal in respiratory acidosis. Metabolic alkalosis frequently occurs in association with other disorders such as respiratory acidosis or alkalosis or metabolic acidosis.
PATHOGENESIS
Metabolic alkalosis occurs as a result of net gain of [HCO3–] or loss of nonvolatile acid (usually HCl by vomiting) from the extracellular fluid. For HCO3– to be added to the extracellular fluid, it must be administered exogenously or synthesized endogenously, in part or entirely by the kidneys. Because it is unusual for alkali to be added to the body, the disorder involves a generative stage, in which the loss of acid usually causes alkalosis, and a maintenance stage, in which the kidneys fail to compensate by excreting HCO3–.
Maintenance of metabolic alkalosis represents a failure of the kidneys to eliminate HCO3– in the usual manner. The kidneys will retain, rather than excrete, the excess alkali and maintain the alkalosis if (1) volume deficiency, chloride deficiency, and K+ deficiency exist in combination with a reduced GFR; or (2) hypokalemia exists because of autonomous hyperaldosteronism. In the first example, alkalosis is corrected by administration of NaCl and KCl, whereas, in the latter, it may be necessary to repair the alkalosis by pharmacologic or surgical intervention, not with saline administration.
DIFFERENTIAL DIAGNOSIS
To establish the cause of metabolic alkalosis (Table 66-6), it is necessary to assess the status of the extracellular fluid volume (ECFV), the recumbent and upright blood pressure, the serum [K+], and the renin-aldosterone system. For example, the presence of chronic hypertension and chronic hypokalemia in an alkalotic patient suggests either mineralocorticoid excess or that the hypertensive patient is receiving diuretics. Low plasma renin activity and normal urine [Na+] and [Cl–] in a patient who is not taking diuretics indicate a primary mineralocorticoid excess syndrome. The combination of hypokalemia and alkalosis in a normotensive, nonedematous patient can be due to Bartter’s or Gitelman’s syndrome, magnesium deficiency, vomiting, exogenous alkali, or diuretic ingestion. Determination of urine electrolytes (especially the urine [Cl–]) and screening of the urine for diuretics may be helpful. If the urine is alkaline, with an elevated [Na+] and [K+] but low [Cl–], the diagnosis is usually either vomiting (overt or surreptitious) or alkali ingestion. If the urine is relatively acid and has low concentrations of Na+, K+, and Cl–, the most likely possibilities are prior vomiting, the posthypercapnic state, or prior diuretic ingestion. If, on the other hand, neither the urine sodium, potassium, nor chloride concentrations are depressed, magnesium deficiency, Bartter’s or Gitelman’s syndrome, or current diuretic ingestion should be considered. Bartter’s syndrome is distinguished from Gitelman’s syndrome because of hypocalciuria and hypomagnesemia in the latter disorder.
CAUSES OF METABOLIC ALKALOSIS |
Abbreviations: DCT, distal convoluted tubule; ECFV, extracellular fluid volume; TALH, thick ascending limb of Henle’s loop.
Alkali Administration Chronic administration of alkali to individuals with normal renal function rarely causes alkalosis. However, in patients with coexistent hemodynamic disturbances, alkalosis can develop because the normal capacity to excrete HCO3– may be exceeded or there may be enhanced reabsorption of HCO3–. Such patients include those who receive HCO3– (PO or IV), acetate loads (parenteral hyperalimentation solutions), citrate loads (transfusions), or antacids plus cation-exchange resins (aluminum hydroxide and sodium polystyrene sulfonate). Nursing home patients receiving tube feedings have a higher incidence of metabolic alkalosis than nursing home patients receiving oral feedings.
METABOLIC ALKALOSIS ASSOCIATED WITH ECFV CONTRACTION, K+ DEPLETION, AND SECONDARY HYPERRENINEMIC HYPERALDOSTERONISM
Gastrointestinal Origin Gastrointestinal loss of H+ from vomiting or gastric aspiration results in retention of HCO3–. During active vomiting, the filtered load of bicarbonate is acutely increased to exceed the reabsorptive capacity of the proximal tubule for HCO3– so that the urine becomes alkaline and high in potassium. When vomiting ceases, the persistence of volume, potassium, and chloride depletion causes maintenance of the alkalosis because of an enhanced capacity of the nephron to reabsorb HCO3–. Correction of the contracted ECFV with NaCl and repair of K+ deficits corrects the acid-base disorder by restoring the ability of the kidney to excrete the excess bicarbonate.
Renal Origin • DIURETICS (See also Chap. 279) Drugs that induce chloruresis, such as thiazides and loop diuretics (furosemide, bumetanide, torsemide, and ethacrynic acid), acutely diminish the ECFV without altering the total body bicarbonate content. The serum [HCO3–] increases because the reduced ECFV “contracts” the [HCO3–] in the plasma (contraction alkalosis). The chronic administration of diuretics tends to generate an alkalosis by increasing distal salt delivery, so that K+ and H+ secretion are stimulated. The alkalosis is maintained by persistence of the contraction of the ECFV, secondary hyperaldosteronism, K+ deficiency, and the direct effect of the diuretic (as long as diuretic administration continues). Repair of the alkalosis is achieved by providing isotonic saline to correct the ECFV deficit.
SOLUTE LOSING DISORDERS: BARTTER’S SYNDROME AND GITELMAN’S SYNDROME See Chap. 339.
NONREABSORBABLE ANIONS AND MAGNESIUM DEFICIENCY Administration of large quantities of nonreabsorbable anions, such as penicillin or carbenicillin, can enhance distal acidification and K+ secretion by increasing the transepithelial potential difference. Mg2+ deficiency results in hypokalemic alkalosis by enhancing distal acidification through stimulation of renin and hence aldosterone secretion.
POTASSIUM DEPLETION Chronic K+ depletion may cause metabolic alkalosis by increasing urinary acid excretion. Both NH4+ production and absorption are enhanced and HCO3– reabsorption is stimulated. Chronic K+ deficiency upregulates the renal H+, K+-ATPase to increase K+ absorption at the expense of enhanced H+ secretion. Alkalosis associated with severe K+ depletion is resistant to salt administration, but repair of the K+ deficiency corrects the alkalosis.
AFTER TREATMENT OF LACTIC ACIDOSIS OR KETOACIDOSIS When an underlying stimulus for the generation of lactic acid or ketoacid is removed rapidly, as with repair of circulatory insufficiency or with insulin therapy, the lactate or ketones are metabolized to yield an equivalent amount of HCO3–. Other sources of new HCO3– are additive with the original amount generated by organic anion metabolism to create a surfeit of HCO3–. Such sources include (1) new HCO3– added to the blood by the kidneys as a result of enhanced acid excretion during the preexisting period of acidosis, and (2) alkali therapy during the treatment phase of the acidosis. Acidosis-induced contraction of the ECFV and K+ deficiency act to sustain the alkalosis.
POSTHYPERCAPNIA Prolonged CO2 retention with chronic respiratory acidosis enhances renal HCO3– absorption and the generation of new HCO3– (increased net acid excretion). Metabolic alkalosis results from the effect of the persistently elevated [HCO3–] when the elevated PaCO2 is abruptly returned toward normal.
METABOLIC ALKALOSIS ASSOCIATED WITH ECFV EXPANSION, HYPERTENSION, AND HYPERALDOSTERONISM
Increased aldosterone levels may be the result of autonomous primary adrenal overproduction or of secondary aldosterone release due to renal overproduction of renin. Mineralocorticoid excess increases net acid excretion and may result in metabolic alkalosis, which may be worsened by associated K+ deficiency. ECFV expansion from salt retention causes hypertension. The kaliuresis persists because of mineralocorticoid excess and distal Na+ absorption causing enhanced K+ excretion, continued K+ depletion with polydipsia, inability to concentrate the urine, and polyuria.
Liddle’s syndrome (Chap. 339) results from increased activity of the collecting duct Na+ channel (ENaC) and is a rare monogenic form of hypertension due to volume expansion manifested as hypokalemic alkalosis and normal aldosterone levels.
Symptoms With metabolic alkalosis, changes in CNS and peripheral nervous system function are similar to those of hypocalcemia (Chap. 423); symptoms include mental confusion; obtundation; and a predisposition to seizures, paresthesia, muscular cramping, tetany, aggravation of arrhythmias, and hypoxemia in chronic obstructive pulmonary disease. Related electrolyte abnormalities include hypokalemia and hypophosphatemia.
RESPIRATORY ACIDOSIS
Respiratory acidosis can be due to severe pulmonary disease, respiratory muscle fatigue, or abnormalities in ventilatory control and is recognized by an increase in PaCO2 and decrease in pH (Table 66-7). In acute respiratory acidosis, there is an immediate compensatory elevation (due to cellular buffering mechanisms) in HCO3–, which increases 1 mmol/L for every 10-mmHg increase in PaCO2. In chronic respiratory acidosis (>24 h), renal adaptation increases the [HCO3–] by 4 mmol/L for every 10-mmHg increase in PaCO2. The serum HCO3– usually does not increase above 38 mmol/L.
RESPIRATORY ACID-BASE DISORDERS |
The clinical features vary according to the severity and duration of the respiratory acidosis, the underlying disease, and whether there is accompanying hypoxemia. A rapid increase in PaCO2 may cause anxiety, dyspnea, confusion, psychosis, and hallucinations and may progress to coma. Lesser degrees of dysfunction in chronic hypercapnia include sleep disturbances; loss of memory; daytime somnolence; personality changes; impairment of coordination; and motor disturbances such as tremor, myoclonic jerks, and asterixis. Headaches and other signs that mimic raised intracranial pressure, such as papilledema, abnormal reflexes, and focal muscle weakness, are due to vasoconstriction secondary to loss of the vasodilator effects of CO2.
Depression of the respiratory center by a variety of drugs, injury, or disease can produce respiratory acidosis. This may occur acutely with general anesthetics, sedatives, and head trauma or chronically with sedatives, alcohol, intracranial tumors, and the syndromes of sleep-disordered breathing including the primary alveolar and obesity-hypoventilation syndromes (Chaps. 318 and 319). Abnormalities or disease in the motor neurons, neuromuscular junction, and skeletal muscle can cause hypoventilation via respiratory muscle fatigue. Mechanical ventilation, when not properly adjusted and supervised, may result in respiratory acidosis, particularly if CO2 production suddenly rises (because of fever, agitation, sepsis, or overfeeding) or alveolar ventilation falls because of worsening pulmonary function. High levels of positive end-expiratory pressure in the presence of reduced cardiac output may cause hypercapnia as a result of large increases in alveolar dead space (Chap. 306e). Permissive hypercapnia is being used with increasing frequency because of studies suggesting lower mortality rates than with conventional mechanical ventilation, especially with severe CNS or heart disease. The respiratory acidosis associated with permissive hypercapnia may require administration of NaHCO3 to increase the arterial pH to 7.25, but overcorrection of the acidemia may be deleterious.
Acute hypercapnia follows sudden occlusion of the upper airway or generalized bronchospasm as in severe asthma, anaphylaxis, inhalational burn, or toxin injury. Chronic hypercapnia and respiratory acidosis occur in end-stage obstructive lung disease. Restrictive disorders involving both the chest wall and the lungs can cause respiratory acidosis because the high metabolic cost of respiration causes ventilatory muscle fatigue. Advanced stages of intrapulmonary and extrapulmonary restrictive defects present as chronic respiratory acidosis.
The diagnosis of respiratory acidosis requires the measurement of PaCO2 and arterial pH. A detailed history and physical examination often indicate the cause. Pulmonary function studies (Chap. 306e), including spirometry, diffusion capacity for carbon monoxide, lung volumes, and arterial PaCO2 and O2 saturation, usually make it possible to determine if respiratory acidosis is secondary to lung disease. The workup for nonpulmonary causes should include a detailed drug history, measurement of hematocrit, and assessment of upper airway, chest wall, pleura, and neuromuscular function.
RESPIRATORY ALKALOSIS
Alveolar hyperventilation decreases PaCO2 and increases the HCO3–/PaCO2 ratio, thus increasing pH (Table 66-7). Nonbicarbonate cellular buffers respond by consuming HCO3–. Hypocapnia develops when a sufficiently strong ventilatory stimulus causes CO2 output in the lungs to exceed its metabolic production by tissues. Plasma pH and [HCO3–] appear to vary proportionately with PaCO2 over a range from 40–15 mmHg. The relationship between arterial [H+] concentration and PaCO2 is ~0.7 mmol/L per mmHg (or 0.01 pH unit/mmHg), and that for plasma [HCO3–] is 0.2 mmol/L per mmHg. Hypocapnia sustained for >2–6 h is further compensated by a decrease in renal ammonium and titratable acid excretion and a reduction in filtered HCO3– reabsorption. Full renal adaptation to respiratory alkalosis may take several days and requires normal volume status and renal function. The kidneys appear to respond directly to the lowered PaCO2 rather than to alkalosis per se. In chronic respiratory alkalosis a 1-mmHg decrease in PaCO2 causes a 0.4-to 0.5-mmol/L drop in [HCO3–] and a 0.3-mmol/L decrease (or 0.003 increase in pH) in [H+].
The effects of respiratory alkalosis vary according to duration and severity but are primarily those of the underlying disease. Reduced cerebral blood flow as a consequence of a rapid decline in PaCO2 may cause dizziness, mental confusion, and seizures, even in the absence of hypoxemia. The cardiovascular effects of acute hypocapnia in the conscious human are generally minimal, but in the anesthetized or mechanically ventilated patient, cardiac output and blood pressure may fall because of the depressant effects of anesthesia and positive-pressure ventilation on heart rate, systemic resistance, and venous return. Cardiac arrhythmias may occur in patients with heart disease as a result of changes in oxygen unloading by blood from a left shift in the hemoglobin-oxygen dissociation curve (Bohr effect). Acute respiratory alkalosis causes intracellular shifts of Na+, K+, and PO42– and reduces free [Ca2+] by increasing the protein-bound fraction. Hypocapnia-induced hypokalemia is usually minor.
Chronic respiratory alkalosis is the most common acid-base disturbance in critically ill patients and, when severe, portends a poor prognosis. Many cardiopulmonary disorders manifest respiratory alkalosis in their early to intermediate stages, and the finding of normocapnia and hypoxemia in a patient with hyperventilation may herald the onset of rapid respiratory failure and should prompt an assessment to determine if the patient is becoming fatigued. Respiratory alkalosis is common during mechanical ventilation.
The hyperventilation syndrome may be disabling. Paresthesia; circumoral numbness; chest wall tightness or pain; dizziness; inability to take an adequate breath; and, rarely, tetany may be sufficiently stressful to perpetuate the disorder. Arterial blood-gas analysis demonstrates an acute or chronic respiratory alkalosis, often with hypocapnia in the range of 15–30 mmHg and no hypoxemia. CNS diseases or injury can produce several patterns of hyperventilation and sustained PaCO2 levels of 20–30 mmHg. Hyperthyroidism, high caloric loads, and exercise raise the basal metabolic rate, but ventilation usually rises in proportion so that arterial blood gases are unchanged and respiratory alkalosis does not develop. Salicylates are the most common cause of drug-induced respiratory alkalosis as a result of direct stimulation of the medullary chemoreceptor (Chap. 472e). The methylxanthines, theophylline, and aminophylline stimulate ventilation and increase the ventilatory response to CO2. Progesterone increases ventilation and lowers arterial PaCO2 by as much as 5–10 mmHg. Therefore, chronic respiratory alkalosis is a common feature of pregnancy. Respiratory alkalosis is also prominent in liver failure, and the severity correlates with the degree of hepatic insufficiency. Respiratory alkalosis is often an early finding in gram-negative septicemia, before fever, hypoxemia, or hypotension develops.
The diagnosis of respiratory alkalosis depends on measurement of arterial pH and PaCO2. The plasma [K+] is often reduced and the [Cl–] increased. In the acute phase, respiratory alkalosis is not associated with increased renal HCO3– excretion, but within hours net acid excretion is reduced. In general, the HCO3– concentration falls by 2.0 mmol/L for each 10-mmHg decrease in PaCO2. Chronic hypocapnia reduces the serum [HCO3–] by 4.0 mmol/L for each 10-mmHg decrease in PaCO2. It is unusual to observe a plasma HCO3– <12 mmol/L as a result of a pure respiratory alkalosis.
When a diagnosis of respiratory alkalosis is made, its cause should be investigated. The diagnosis of hyperventilation syndrome is made by exclusion. In difficult cases, it may be important to rule out other conditions such as pulmonary embolism, coronary artery disease, and hyperthyroidism.
SECTION 8 | ALTERATIONS IN SEXUAL FUNCTION AND REPRODUCTION |
67 | Sexual Dysfunction |
Male sexual dysfunction affects 10–25% of middle-aged and elderly men, and female sexual dysfunction occurs with similar frequency. Demographic changes, the popularity of newer treatments, and greater awareness of sexual dysfunction by patients and society have led to increased diagnosis and associated health care expenditures for the management of this common disorder. Because many patients are reluctant to initiate discussion of their sex lives, physicians should address this topic directly to elicit a history of sexual dysfunction.
MALE SEXUAL DYSFUNCTION
PHYSIOLOGY OF MALE SEXUAL RESPONSE
Normal male sexual function requires (1) an intact libido, (2) the ability to achieve and maintain penile erection, (3) ejaculation, and (4) detumescence. Libido refers to sexual desire and is influenced by a variety of visual, olfactory, tactile, auditory, imaginative, and hormonal stimuli. Sex steroids, particularly testosterone, act to increase libido. Libido can be diminished by hormonal or psychiatric disorders and by medications.
Penile tumescence leading to erection depends on an increased flow of blood into the lacunar network accompanied by complete relaxation of the arteries and corporal smooth muscle. The microarchitecture of the corpora is composed of a mass of smooth muscle (trabecula) that contains a network of endothelial-lined vessels (lacunar spaces). Subsequent compression of the trabecular smooth muscle against the fibroelastic tunica albuginea causes a passive closure of the emissary veins and accumulation of blood in the corpora. In the presence of a full erection and a competent valve mechanism, the corpora become noncompressible cylinders from which blood does not escape.
The central nervous system (CNS) exerts an important influence by either stimulating or antagonizing spinal pathways that mediate erectile function and ejaculation. The erectile response is mediated by a combination of central (psychogenic) innervation and peripheral (reflexogenic) innervation. Sensory nerves that originate from receptors in the penile skin and glans converge to form the dorsal nerve of the penis, which travels to the S2-S4 dorsal root ganglia via the pudendal nerve. Parasympathetic nerve fibers to the penis arise from neurons in the intermediolateral columns of the S2-S4 sacral spinal segments. Sympathetic innervation originates from the T11 to the L2 spinal segments and descends through the hypogastric plexus.
Neural input to smooth-muscle tone is crucial to the initiation and maintenance of an erection. There is also an intricate interaction between the corporal smooth-muscle cell and its overlying endothelial cell lining (Fig. 67-1). Nitric oxide, which induces vascular relaxation, promotes erection and is opposed by endothelin 1 (ET-1) and Rho kinase, which mediate vascular contraction. Nitric oxide is synthesized from l-arginine by nitric oxide synthase and is released from the nonadrenergic, noncholinergic (NANC) autonomic nerve supply to act postjunctionally on smooth-muscle cells. Nitric oxide increases the production of cyclic 3′,5′-guanosine monophosphate (cyclic GMP), which induces relaxation of smooth muscle (Fig. 67-2). Cyclic GMP is gradually broken down by phosphodiesterase type 5 (PDE-5). Inhibitors of PDE-5, such as the oral medications sildenafil, vardenafil, and tadalafil, maintain erections by reducing the breakdown of cyclic GMP. However, if nitric oxide is not produced at some level, PDE-5 inhibitors are ineffective, as these drugs facilitate, but do not initiate, the initial enzyme cascade. In addition to nitric oxide, vasoactive prostaglandins (PGE1, PGF2α) are synthesized within the cavernosal tissue and increase cyclic AMP levels, also leading to relaxation of cavernosal smooth-muscle cells.
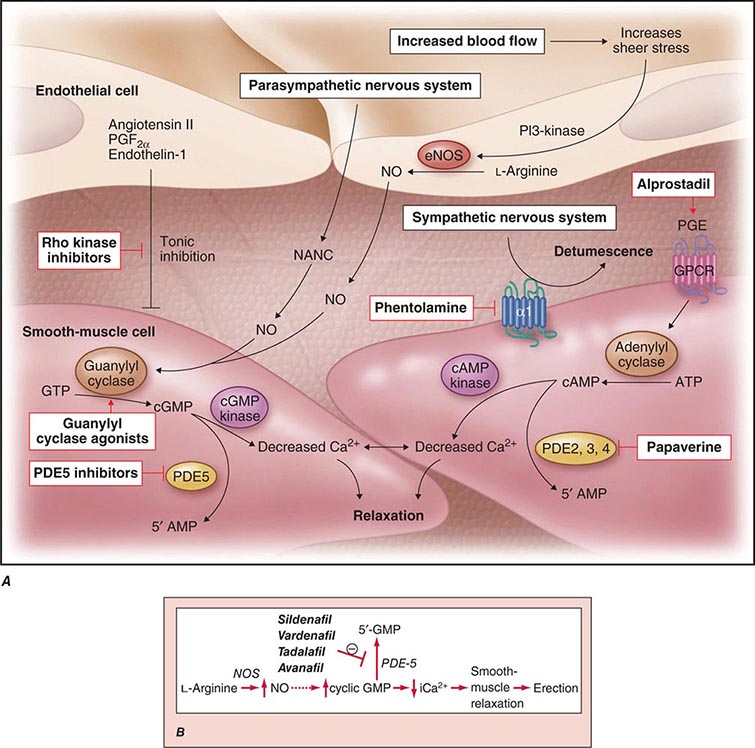
FIGURE 67-1 Pathways that regulate penile smooth-muscle relaxation and erection. A. Outflow from the parasympathetic nervous system leads to relaxation of the cavernous sinusoids in two ways, both of which increase the concentration of nitric oxide (NO) in smooth-muscle cells. First, NO is the neurotransmitter in nonadrenergic, noncholinergic (NANC) fibers; second, stimulation of endothelial nitric oxide synthase (eNOS) through cholinergic output causes increased production of NO. The NO produced in the endothelium then diffuses into the smooth-muscle cells and decreases its intracellular calcium concentration through a pathway mediated by cyclic guanosine monophosphate (cGMP), leading to relaxation. A separate mechanism that decreases the intracellular calcium level is mediated by cyclic adenosine monophosphate (cAMP). With increased cavernosal blood flow, as well as increased levels of vascular endothelial growth factor (VEGF), the endothelial release of NO is further sustained through the phosphatidylinositol 3 (PI3) kinase pathway. Active treatments (red boxes) include drugs that affect the cGMP pathway (phosphodiesterase [PDE] type 5 inhibitors and guanylyl cyclase agonists), the cAMP pathway (alprostadil), or both pathways (papaverine), along with neural-tone mediators (phentolamine and Rho kinase inhibitors). Agents that are being developed include guanylyl cyclase agonists (to bypass the need for endogenous NO) and Rho kinase inhibitors (to inhibit tonic contraction of smooth-muscle cells mediated through endothelin). α1, α-adrenergic receptor; GPCR, G-protein–coupled receptor, GTP, guanosine triphosphate; PGE, prostaglandin E; PGF, prostaglandin F. B. Biochemical pathways of NO synthesis and action. Sildenafil, vardenafil, and tadalafil enhance erectile function by inhibiting phosphodiesterase type 5 (PDE-5), thereby maintaining high levels of cyclic 3′,5′-guanosine monophosphate (cyclic GMP). iCa2+, intracellular calcium; NOS, nitric oxide synthase. (Part A from K McVary: N Engl J Med 357:2472, 2007; with permission.)
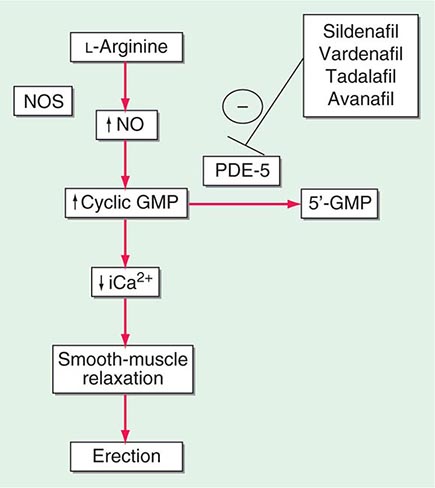
FIGURE 67-2 Biochemical pathways modified by phosphodiesterase type 5 (PDE-5) inhibitors. Sildenafil, vardenafil, tadalafil and avanafil enhance erectile function by inhibiting PDE-5, thereby maintaining high levels of cyclic 3′,5′-guanosine monophosphate (cyclic GMP). iCa2+, intracellular calcium; NO, nitric oxide; NOS, nitric oxide synthase.
Ejaculation is stimulated by the sympathetic nervous system; this results in contraction of the epididymis, vas deferens, seminal vesicles, and prostate, causing seminal fluid to enter the urethra. Seminal fluid emission is followed by rhythmic contractions of the bulbocavernosus and ischiocavernosus muscles, leading to ejaculation. Premature ejaculation usually is related to anxiety or a learned behavior and is amenable to behavioral therapy or treatment with medications such as selective serotonin reuptake inhibitors (SSRIs). Retrograde ejaculation results when the internal urethral sphincter does not close; it may occur in men with diabetes or after surgery involving the bladder neck.
Detumescence is mediated by norepinephrine from the sympathetic nerves, endothelin from the vascular surface, and smooth-muscle contraction induced by postsynaptic α-adrenergic receptors and activation of Rho kinase. These events increase venous outflow and restore the flaccid state. Venous leak can cause premature detumescence and is caused by insufficient relaxation of the corporal smooth muscle rather than a specific anatomic defect. Priapism refers to a persistent and painful erection and may be associated with sickle cell anemia, hypercoagulable states, spinal cord injury, or injection of vasodilator agents into the penis.
ERECTILE DYSFUNCTION
Epidemiology Erectile dysfunction (ED) is not considered a normal part of the aging process. Nonetheless, it is associated with certain physiologic and psychological changes related to age. In the Massachusetts Male Aging Study (MMAS), a community-based survey of men age 40–70, 52% of responders reported some degree of ED. Complete ED occurred in 10% of respondents, moderate ED in 25%, and minimal ED in 17%. The incidence of moderate or severe ED more than doubled between the ages of 40 and 70. In the National Health and Social Life Survey (NHSLS), which included a sample of men and women age 18–59, 10% of men reported being unable to maintain an erection (corresponding to the proportion of men in the MMAS reporting severe ED). Incidence was highest among men in the age group 50–59 (21%) and men who were poor (14%), divorced (14%), and less educated (13%).
The incidence of ED is also higher among men with certain medical disorders, such as diabetes mellitus, obesity, lower urinary tract symptoms secondary to benign prostatic hyperplasia (BPH), heart disease, hypertension, decreased high-density lipoprotein (HDL) levels, and diseases associated with general systemic inflammation (e.g., rheumatoid arthritis). Cardiovascular disease and ED share etiologies as well as pathophysiology (e.g., endothelial dysfunction), and the degree of ED appears to correlate with the severity of cardiovascular disease. Consequently, ED represents a “sentinel symptom” in patients with occult cardiovascular and peripheral vascular disease.
Smoking is also a significant risk factor in the development of ED. Medications used in treating diabetes or cardiovascular disease are additional risk factors (see below). There is a higher incidence of ED among men who have undergone radiation or surgery for prostate cancer and in those with a lower spinal cord injury. Psychological causes of ED include depression, anger, stress from unemployment, and other stress-related causes.
Pathophysiology ED may result from three basic mechanisms: (1) failure to initiate (psychogenic, endocrinologic, or neurogenic), (2) failure to fill (arteriogenic), and (3) failure to store adequate blood volume within the lacunar network (venoocclusive dysfunction). These categories are not mutually exclusive, and multiple factors contribute to ED in many patients. For example, diminished filling pressure can lead secondarily to venous leak. Psychogenic factors frequently coexist with other etiologic factors and should be considered in all cases. Diabetic, atherosclerotic, and drug-related causes account for >80% of cases of ED in older men.
VASCULOGENIC The most common organic cause of ED is a disturbance of blood flow to and from the penis. Atherosclerotic or traumatic arterial disease can decrease flow to the lacunar spaces, resulting in decreased rigidity and an increased time to full erection. Excessive outflow through the veins despite adequate inflow also may contribute to ED. Structural alterations to the fibroelastic components of the corpora may cause a loss of compliance and inability to compress the tunical veins. This condition may result from aging, increased cross-linking of collagen fibers induced by nonenzymatic glycosylation, hypoxemia, or altered synthesis of collagen associated with hypercholesterolemia.
NEUROGENIC Disorders that affect the sacral spinal cord or the autonomic fibers to the penis preclude nervous system relaxation of penile smooth muscle, thus leading to ED. In patients with spinal cord injury, the degree of ED depends on the completeness and level of the lesion. Patients with incomplete lesions or injuries to the upper part of the spinal cord are more likely to retain erectile capabilities than are those with complete lesions or injuries to the lower part. Although 75% of patients with spinal cord injuries have some erectile capability, only 25% have erections sufficient for penetration. Other neurologic disorders commonly associated with ED include multiple sclerosis and peripheral neuropathy. The latter is often due to either diabetes or alcoholism. Pelvic surgery may cause ED through disruption of the autonomic nerve supply.
ENDOCRINOLOGIC Androgens increase libido, but their exact role in erectile function is unclear. Individuals with castrate levels of testosterone can achieve erections from visual or sexual stimuli. Nonetheless, normal levels of testosterone appear to be important for erectile function, particularly in older males. Androgen replacement therapy can improve depressed erectile function when it is secondary to hypogonadism; however, it is not useful for ED when endogenous testosterone levels are normal. Increased prolactin may decrease libido by suppressing gonadotropin-releasing hormone (GnRH), and it also leads to decreased testosterone levels. Treatment of hyperprolactinemia with dopamine agonists can restore libido and testosterone.
DIABETIC ED occurs in 35–75% of men with diabetes mellitus. Pathologic mechanisms are related primarily to diabetes-associated vascular and neurologic complications. Diabetic macrovascular complications are related mainly to age, whereas microvascular complications correlate with the duration of diabetes and the degree of glycemic control (Chap. 417). Individuals with diabetes also have reduced amounts of nitric oxide synthase in both endothelial and neural tissues.
PSYCHOGENIC Two mechanisms contribute to the inhibition of erections in psychogenic ED. First, psychogenic stimuli to the sacral cord may inhibit reflexogenic responses, thereby blocking activation of vasodilator outflow to the penis. Second, excess sympathetic stimulation in an anxious man may increase penile smooth-muscle tone. The most common causes of psychogenic ED are performance anxiety, depression, relationship conflict, loss of attraction, sexual inhibition, conflicts over sexual preference, sexual abuse in childhood, and fear of pregnancy or sexually transmitted disease. Almost all patients with ED, even when it has a clear-cut organic basis, develop a psychogenic component as a reaction to ED.
MEDICATION-RELATED Medication-induced ED (Table 67-1) is estimated to occur in 25% of men seen in general medical outpatient clinics. The adverse effects related to drug therapy are additive, especially in older men. In addition to the drug itself, the disease being treated is likely to contribute to sexual dysfunction. Among the antihypertensive agents, the thiazide diuretics and beta blockers have been implicated most frequently. Calcium channel blockers and angiotensin converting-enzyme inhibitors are cited less frequently. These drugs may act directly at the corporal level (e.g., calcium channel blockers) or indirectly by reducing pelvic blood pressure, which is important in the development of penile rigidity. α-Adrenergic blockers are less likely to cause ED. Estrogens, GnRH agonists, H2 antagonists, and spironolactone cause ED by suppressing gonadotropin production or by blocking androgen action. Antidepressant and antipsychotic agents—particularly neuroleptics, tricyclics, and SSRIs—are associated with erectile, ejaculatory, orgasmic, and sexual desire difficulties.
DRUGS ASSOCIATED WITH ERECTILE DYSFUNCTION |
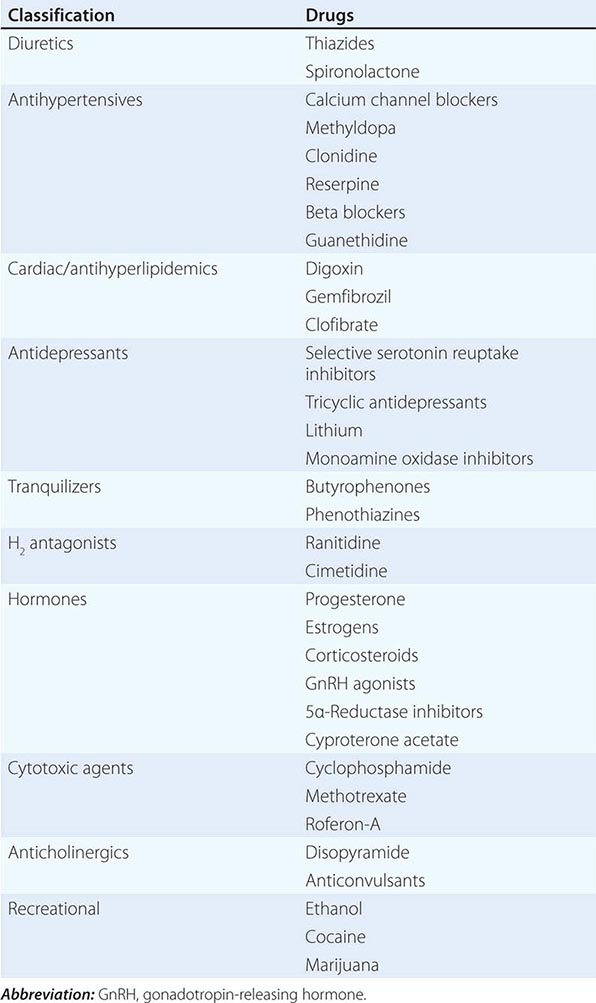
If there is a strong association between the institution of a drug and the onset of ED, alternative medications should be considered. Otherwise, it is often practical to treat the ED without attempting multiple changes in medications, as it may be difficult to establish a causal role for a drug.
APPROACH TO THE PATIENT:
Erectile Dysfunction
A good physician-patient relationship helps unravel the possible causes of ED, many of which require discussion of personal and sometimes embarrassing topics. For this reason, a primary care provider is often ideally suited to initiate the evaluation. However, a significant percentage of men experience ED and remain undiagnosed unless specifically questioned about this issue. By far the two most common reasons for underreporting of ED are patient embarrassment and perceptions of physicians’ inattention to the disease. Once the topic is initiated by the physician, patients are more willing to discuss their potency issues. A complete medical and sexual history should be taken in an effort to assess whether the cause of ED is organic, psychogenic, or multifactorial (Fig. 67-3).
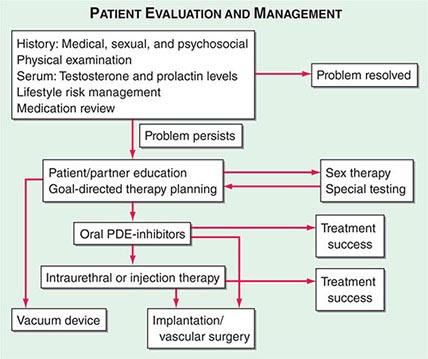
FIGURE 67-3 Algorithm for the evaluation and management of patients with erectile dysfunction. PDE, phosphodiesterase.
Both the patient and his sexual partner should be interviewed regarding sexual history. ED should be distinguished from other sexual problems, such as premature ejaculation. Lifestyle factors such as sexual orientation, the patient’s distress from ED, performance anxiety, and details of sexual techniques should be addressed. Standardized questionnaires are available to assess ED, including the International Index of Erectile Function (IIEF) and the more easily administered Sexual Health Inventory for Men (SHIM), a validated abridged version of the IIEF.
The initial evaluation of ED begins with a review of the patient’s medical, surgical, sexual, and psychosocial histories. The history should note whether the patient has experienced pelvic trauma, surgery, or radiation. In light of the increasing recognition of the relationship between lower urinary tract symptoms and ED, it is advisable to evaluate for the presence of symptoms of bladder outlet obstruction. Questions should focus on the onset of symptoms, the presence and duration of partial erections, and the progression of ED. A history of nocturnal or early morning erections is useful for distinguishing physiologic ED from psychogenic ED. Nocturnal erections occur during rapid eye movement (REM) sleep and require intact neurologic and circulatory systems. Organic causes of ED generally are characterized by a gradual and persistent change in rigidity or the inability to sustain nocturnal, coital, or self-stimulated erections. The patient should be questioned about the presence of penile curvature or pain with coitus. It is also important to address libido, as decreased sexual drive and ED are sometimes the earliest signs of endocrine abnormalities (e.g., increased prolactin, decreased testosterone levels). It is useful to ask whether the problem is confined to coitus with one partner or also involves other partners; ED not uncommonly arises in association with new or extramarital sexual relationships. Situational ED, as opposed to consistent ED, suggests psychogenic causes. Ejaculation is much less commonly affected than erection, but questions should be asked about whether ejaculation is normal, premature, delayed, or absent. Relevant risk factors should be identified, such as diabetes mellitus, coronary artery disease (CAD), and neurologic disorders. The patient’s surgical history should be explored with an emphasis on bowel, bladder, prostate, and vascular procedures. A complete drug history is also important. Social changes that may precipitate ED are also crucial to the evaluation, including health worries, spousal death, divorce, relationship difficulties, and financial concerns.
Because ED commonly involves a host of endothelial cell risk factors, men with ED report higher rates of overt and silent myocardial infarction. Therefore, ED in an otherwise asymptomatic male warrants consideration of other vascular disorders, including CAD.
The physical examination is an essential element in the assessment of ED. Signs of hypertension as well as evidence of thyroid, hepatic, hematologic, cardiovascular, or renal diseases should be sought. An assessment should be made of the endocrine and vascular systems, the external genitalia, and the prostate gland. The penis should be palpated carefully along the corpora to detect fibrotic plaques. Reduced testicular size and loss of secondary sexual characteristics are suggestive of hypogonadism. Neurologic examination should include assessment of anal sphincter tone, investigation of the bulbocavernosus reflex, and testing for peripheral neuropathy.
Although hyperprolactinemia is uncommon, a serum prolactin level should be measured, as decreased libido and/or ED may be the presenting symptoms of a prolactinoma or another mass lesion of the sella (Chap. 403). The serum testosterone level should be measured, and if it is low, gonadotropins should be measured to determine whether hypogonadism is primary (testicular) or secondary (hypothalamic-pituitary) in origin (Chap. 411). If not performed recently, serum chemistries, complete blood count (CBC), and lipid profiles may be of value, as they can yield evidence of anemia, diabetes, hyperlipidemia, or other systemic diseases associated with ED. Determination of serum prostate-specific antigen (PSA) should be conducted according to recommended clinical guidelines (Chap. 115).
Additional diagnostic testing is rarely necessary in the evaluation of ED. However, in selected patients, specialized testing may provide insight into pathologic mechanisms of ED and aid in the selection of treatment options. Optional specialized testing includes (1) studies of nocturnal penile tumescence and rigidity, (2) vascular testing (in-office injection of vasoactive substances, penile Doppler ultrasound, penile angiography, dynamic infusion cavernosography/cavernosometry), (3) neurologic testing (biothesiometry-graded vibratory perception, somatosensory evoked potentials), and (4) psychological diagnostic tests. The information potentially gained from these procedures must be balanced against their invasiveness and cost.
TREATMENT | MALE SEXUAL DYSFUNCTION |
PATIENT EDUCATION
Patient and partner education is essential in the treatment of ED. In goal-directed therapy, education facilitates understanding of the disease, the results of the tests, and the selection of treatment. Discussion of treatment options helps clarify how treatment is best offered and stratify first- and second-line therapies. Patients with high-risk lifestyle issues such as obesity, smoking, alcohol abuse, and recreational drug use should be counseled on the role those factors play in the development of ED.
Therapies currently employed for the treatment of ED include oral PDE-5 inhibitor therapy (most commonly used), injection therapies, testosterone therapy, penile devices, and psychological therapy. In addition, limited data suggest that treatments for underlying risk factors and comorbidities—for example, weight loss, exercise, stress reduction, and smoking cessation—may improve erectile function. Decisions regarding therapy should take into account the preferences and expectations of patients and their partners.
ORAL AGENTS
Sildenafil, tadalafil, vardenafil, and avanafil are the only approved and effective oral agents for the treatment of ED. These four medications have markedly improved the management of ED because they are effective for the treatment of a broad range of causes, including psychogenic, diabetic, vasculogenic, post-radical prostatectomy (nerve-sparing procedures), and spinal cord injury. They belong to a class of medications that are selective and potent inhibitors of PDE-5, the predominant phosphodiesterase isoform found in the penis. They are administered in graduated doses and enhance erections after sexual stimulation. The onset of action is approximately 30–120 min, depending on the medication used and other factors, such as recent food intake. Reduced initial doses should be considered for patients who are elderly, are taking concomitant alpha blockers, have renal insufficiency, or are taking medications that inhibit the CYP3A4 metabolic pathway in the liver (e.g., erythromycin, cimetidine, ketoconazole, and possibly itraconazole and mibefradil), as they may increase the serum concentration of the PDE-5 inhibitors (PDE-5i) or promote hypotension.
Initially, there were concerns about the cardiovascular safety of PDE-5i drugs. These agents can act as a mild vasodilator, and warnings exist about orthostatic hypotension with concomitant use of alpha blockers. The use of PDE-5i is not contraindicated in men who are also receiving alpha blockers, but they must be stabilized on this blood pressure medication prior to initiating therapy. Concerns also existed that use of PDE-5i would increase cardiovascular events. However, the safety of these drugs has been confirmed in several controlled trials with no increase in myocardial ischemic events or overall mortality compared to the general population.
Several randomized trials have demonstrated the efficacy of this class of medications. There are no compelling data to support the superiority of one PDE-5i over another. Subtle differences between agents have variable clinical relevance (Table 67-2).
CHARACTERISTICS OF PDE-5i MEDICATIONS |
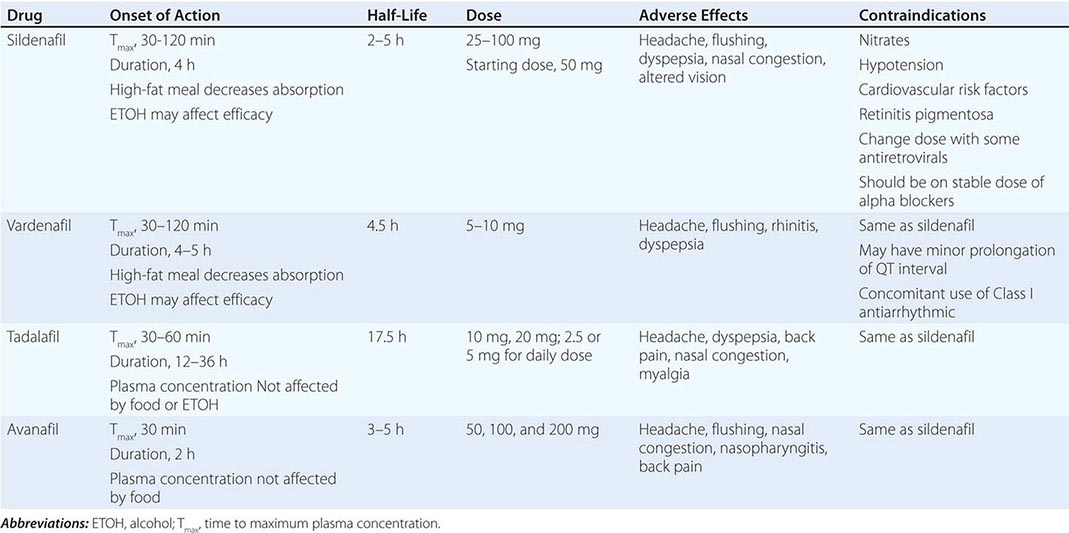
Patients may fail to respond to a PDE-5i for several reasons (Table 67-3). Some patients may not tolerate PDE-5i secondary to adverse events from vasodilation in nonpenile tissues expressing PDE-5 or from the inhibition of homologous nonpenile isozymes (i.e., PDE-6 found in the retina). Abnormal vision attributed to the effects of PDE-5i on retinal PDE-6 is of short duration, reported only with sildenafil and not thought to be clinically significant. A more serious concern is the possibility that PDE-5i may cause nonarteritic anterior ischemic optic neuropathy; although data to support that association are limited, it is prudent to avoid the use of these agents in men with a prior history of nonarteritic anterior ischemic optic neuropathy.
ISSUES TO CONSIDER IF PATIENTS REPORT FAILURE OF PDE-5i TO IMPROVE ERECTILE DYSFUNCTION |
Abbreviations: NO, nitric oxide; PDE-5i, phosphodiesterase type 5 inhibitor.
Testosterone supplementation combined with a PDE-5i may be beneficial in improving erectile function in hypogonadal men with ED who are unresponsive to PDE-5i alone. These drugs do not affect ejaculation, orgasm, or sexual drive. Side effects associated with PDE-5i include headaches (19%), facial flushing (9%), dyspepsia (6%), and nasal congestion (4%). Approximately 7% of men using sildenafil may experience transient altered color vision (blue halo effect), and 6% of men taking tadalafil may experience loin pain. PDE-5i is contraindicated in men receiving nitrate therapy for cardiovascular disease, including agents delivered by the oral, sublingual, transnasal, and topical routes. These agents can potentiate its hypotensive effect and may result in profound shock. Likewise, amyl/butyl nitrate “poppers” may have a fatal synergistic effect on blood pressure. PDE-5i also should be avoided in patients with congestive heart failure and cardiomyopathy because of the risk of vascular collapse. Because sexual activity leads to an increase in physiologic expenditure (5–6 metabolic equivalents [METS]), physicians have been advised to exercise caution in prescribing any drug for sexual activity to those with active coronary disease, heart failure, borderline hypotension, or hypovolemia and to those on complex antihypertensive regimens.
Although the various forms of PDE-5i have a common mechanism of action, there are a few differences among the four agents (Table 67-2). Tadalafil is unique in its longer half-life, whereas avanafil appears to have the most rapid onset of action. All four drugs are effective for patients with ED of all ages, severities, and etiologies. Although there are pharmacokinetic and pharmacodynamic differences among these agents, clinically relevant differences are not clear.
ANDROGEN THERAPY
Testosterone replacement is used to treat both primary and secondary causes of hypogonadism (Chap. 411). Androgen supplementation in the setting of normal testosterone is rarely efficacious in the treatment of ED and is discouraged. Methods of androgen replacement include transdermal patches and gels, parenteral administration of long-acting testosterone esters (enanthate and cypionate), and oral preparations (17 α-alkylated derivatives) (Chap. 411). Oral androgen preparations have the potential for hepatotoxicity and should be avoided.
Men who receive testosterone should be reevaluated after 1–3 months and at least annually thereafter for testosterone levels, erectile function, and adverse effects, which may include gynecomastia, sleep apnea, development or exacerbation of lower urinary tract symptoms or BPH, prostate cancer, lowering of HDL, erythrocytosis, elevations of liver function tests, and reduced fertility. Periodic reevaluation should include measurement of CBC and PSA and digital rectal exam. Therapy should be discontinued in patients who do not respond within 3 months.
VACUUM CONSTRICTION DEVICES
Vacuum constriction devices (VCDs) are a well-established noninvasive therapy. They are a reasonable treatment alternative for select patients who cannot take sildenafil or do not desire other interventions. VCDs draw venous blood into the penis and use a constriction ring to restrict venous return and maintain tumescence. Adverse events with VCD include pain, numbness, bruising, and altered ejaculation. Additionally, many patients complain that the devices are cumbersome and that the induced erections have a nonphysiologic appearance and feel.
INTRAURETHRAL ALPROSTADIL
If a patient fails to respond to oral agents, a reasonable next choice is intraurethral or self-injection of vasoactive substances. Intraurethral prostaglandin E1 (alprostadil), in the form of a semisolid pellet (doses of 125–1000 μg), is delivered with an applicator. Approximately 65% of men receiving intraurethral alprostadil respond with an erection when tested in the office, but only 50% achieve successful coitus at home. Intraurethral insertion is associated with a markedly reduced incidence of priapism in comparison to intracavernosal injection.
INTRACAVERNOSAL SELF-INJECTION
Injection of synthetic formulations of alprostadil is effective in 70–80% of patients with ED, but discontinuation rates are high because of the invasive nature of administration. Doses range between 1 and 40 μg. Injection therapy is contraindicated in men with a history of hypersensitivity to the drug and men at risk for priapism (hypercoagulable states, sickle cell disease). Side effects include local adverse events, prolonged erections, pain, and fibrosis with chronic use. Various combinations of alprostadil, phentolamine, and/or papaverine sometimes are used.
SURGERY
A less frequently used form of therapy for ED involves the surgical implantation of a semirigid or inflatable penile prosthesis. The choice of prosthesis is dependent on patient preference and should take into account body habitus and manual dexterity, which may affect the ability of the patient to manipulate the device. Because of the permanence of prosthetic devices, patients should be advised to first consider less invasive options for treatment. These surgical treatments are invasive, are associated with potential complications, and generally are reserved for treatment of refractory ED. Despite their high cost and invasiveness, penile prostheses are associated with high rates of patient and partner satisfaction.
SEX THERAPY
A course of sex therapy may be useful for addressing specific interpersonal factors that may affect sexual functioning. Sex therapy generally consists of in-session discussion and at-home exercises specific to the person and the relationship. Psychosexual therapy involves techniques such as sensate focus (nongenital massage), sensory awareness exercises, correction of misconceptions about sexuality, and interpersonal difficulties therapy (e.g., open communication about sexual issues, physical intimacy scheduling, and behavioral interventions). These approaches may be useful in patients who have psychogenic or social components to their ED, although data from randomized trials are scanty and inconsistent. It is preferable if therapy includes both partners if the patient is involved in an ongoing relationship.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


