Fig. 3.1
Schematic representation of the fate of topically applied drugs on the eye and their disposition
3.2.2 Pre-corneal Factors Affecting Ocular Penetration of Drugs after Topical Application
In the air-corneal interface, one would see several layers of physiological importance involved in the maintenance of corneal integrity and transparency very efficiently. The outer most layer is a mono lipid layer floating on the tear film having hydrophobic lipids toward air and amphiphilic lipids in the lipid-water interface thereby giving integrity. These lipids are secreted by meibomian glands, which are embedded in the tarsal plate of the upper and lower eyelids giving protection against aqueous evaporation. The cornea produces a small proportion of the aqueous layer as well as mucins in the glycocalyx. The conjunctiva secretes substantial electrolytes and water into the aqueous layer and mucins into the mucous layer. At the same time, the conjunctiva can also absorb electrolytes, water, and applied drugs from the tear film thereby modifying their pharmacokinetics. Although five layers of the cornea are very distinct, the corneal epithelium, stroma, and endothelium are the layers that gain pharmacological importance as far as drug penetration is concerned. In these layers, drug transport across the cornea can be modulated by drug transporters present in the epithelium and endothelium. However, the resistance for the transport of the drug across these membranes is also affected by the physiochemical nature of the molecule. The corneal epithelium is hydrophobic and the stroma is hydrophilic; thereby both water-soluble and lipid-soluble drugs cannot penetrate freely. The cornea behaves as a typical biological membrane in which most of the drugs cross this structure either by intracellular or transcellular diffusion (Lee and Robinson 1986).
Topically applied eyedrops exceeding the volume of 20 µl would be drained from the cul-de-sac via nasolacrimal duct or removed externally by blinking. Application of low volumes of eyedrop would be beneficial for getting better bioavailability. Volume of solution delivered by commercial eye droppers can be between 25–50 µl and if the subject is not blinking, eye can hold upto 30 µl if instilled carefully. However, reflex blinking may increase both solution drainage and overflow from the conjunctival sac. Decreasing the volume of instillation is expected to improve risk-to-benefit ratio of the potent compounds known to have systemic toxicity (Lynch et al. 1987).
Most of the topically applied drugs are the salts of acids or bases like ciprofloxacin HCl, timolol maleate, prednisolone acetate, pilocarpine trinitrate, atropine sulfate, bromfenac sodium, etc. Drug molecules in the tear film can be influenced or get influenced by the tear film pH depending on their concentration and property. Therefore, the extent of their ocular bioavailability would depend upon their ionization constant (pKa) under the given pH of the pre-corneal tear film. The unionized compounds show higher lipophilicity enabling them to cross the outer epithelium. Apart from pKa, molecular weight, preservatives, presence of surfactants, vehicle, and osmolarity of the formulation are the other factors that determine the ocular pharmacokinetics of the drugs. Certain topical drug preservatives like benzalkonium chloride are reported to increase the ocular bioavailability of the topically applied drugs by disrupting the corneal membrane.
Most of the topically instilled drugs reach their Cmax in aqueous humor between 15 min to 2 hrs. Based on the frequency of drug administration hourly or 2 hourly, the levels can substantially be increased to reach pharmacologically accepted concentration in the cornea, conjunctiva, and aqueous humor. Pre-corneal drug residence time is largely dependent upon the bidirectional equilibration of drug levels reaching in between the cornea and tear film which slowly decrease along with time. This aspect gains much importance when trying to reach adequate antimicrobial drug levels after repeated drug instillation in case of corneal or conjunctival bacterial infections. Corneal nerves are very sensitive to non-specific stimulation, pH and osmolarity, the pre-corneal elimination of drugs which is in equilibrium with tissues can be accelerated in case if the lacrimal secretion is increased by the stimulation of sensory nerves of the cornea due to the nature of the drug component or formulation factors. Composition of topical formulation (drug vehicle) has also been reported to influence the topical penetration of drugs (Hardberger et al. 1975; McCarthy 1975).
3.2.3 Compartment Models Used to Predict Ocular Kinetics of Drugs
Compartment models are mathematical equations used to predict the drug disposition beyond the tissues/fluids in which the drug concentration is quantified. This information is required to understand the movement of drugs across various tissues and organs along with the elimination of drugs from the primary compartment. Usually for systemic pharmacokinetics, blood would serve as a central compartment from where the drug elimination predominately happens. In eye, anterior chamber administration of amikacin and chloramphenicol followed one compartmental model (Mayers et al. 1991). Following bolus administration of the hydrophobic immune-suppressant drug cyclosporine into the anterior chamber, its clearance from the aqueous humor was predicted by one-compartment model with the terminal half-life of 30–40 h, where as for the aqueous to tissue distribution into the cornea, a two-compartment model was employed (Oh et al. 1995).
Typically, when the analysis of drug partition into systemic and ocular tissues needs to be done, two-compartment model best suited the purpose considering that the multiple sites of tissue distribution is involved. Two-compartment modeling was successfully used by several investigators to predict the drug partitioning from tear film to ocular tissues for their effect as well as systemic absorption causing side effects. The advantage of timolol with thickening agent (gel) was compared with simple topical solution of timolol in rabbits. A two-compartment pharmacokinetic model was used to fit the aqueous humor level for determining the drainage (kd) and absorption rate constants (ka) in the pre-corneal area as well as the elimination rate constant (ke) of timolol in the aqueous humor. This study reported that gel has a longer retention time in eyes to improve ocular bioavailability and lesser systemic side effects (Chiang et al. 1996). Sasaki and coworkers (2000) predicted concentrations of tilisolol in the aqueous humor after instillation with CMC vehicle from the tear concentrations. Its ocular and systemic absorption was analyzed by a mathematical model including a diffusion process and a two-compartment model with first-order absorption, respectively. Similarly, a two-compartment model was successfully used for topically instilled gentamicin (Eljarrat‐Binstock et al. 2004) and chlorhexidine gluconate (Xuguang et al. 2006). Presence of drug transporters is well recognized in the blood-ocular barriers; therefore, systemically administered drugs partitioning into the humors or tissues are subjected to their susceptibility for drug influx/efflux mechanisms. Systemically administered P-gp substrate (Rho-123) was best fitted with a three-compartment model and shifted to a two-compartment model upon its blocker administration in rabbits. This shift has increased intraocular penetration of compounds across blood-ocular barriers (Senthilkumari et al. 2008a, b). A three-compartment physiological-based pharmacokinetic (PK) model with a bidirectional transfer between the cornea and aqueous humor and a unidirectional transfer between the aqueous humor and iris-ciliary body was used to describe an antiglaucoma agent 1-ethyl-6-fluoro,1,2,3,4-tetrahydroquinoline in rabbits. Using its ED50 values derived from pressure-lowering activity (pharmacodynamic property), the PK-PD model was derived to explain pharmacodynamics of iris-ciliary body concentration time data (Pamulapati and Schoenwald 2012).
Intravitreally administered drugs reaching the systemic circulation is of concern for their toxicity (Fig. 3.2). Systemic pharmacokinetics of intravitreally administered ranibizumab in patients with retinal vein occlusion (RVO) or diabetic macular edema (DME) was predicted using one-compartment pharmacokinetics model with first-order absorption and elimination rate constants. This population kinetic study found that there is no difference among conditions like AMD, RVO, and DME as far as the systemic exposure of VEGF antibody is concerned (Zhang et al. 2014).
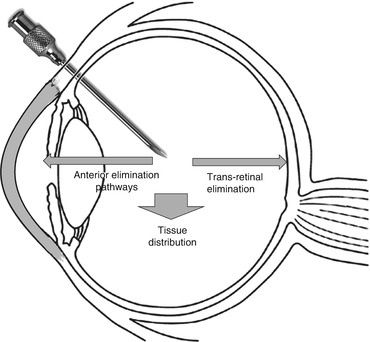
Fig. 3.2
Elimination pathways of intravitreally injected drugs
Subconjunctival injection of gentamicin was fitted into one compartmental model (van Rooyen et al. 1991). Antibacterial agents such as ciprofloxacin and fleroxacin (Miller et al. 1992) were also predicted with single-compartment model after direct intravitreal administration. A population pharmacokinetic metabolism model was used to describe the concentrations of ciprofloxacin in serum, aqueous and vitreous humor by a four-compartment PK linear model after oral administration.
Single-dose administration as eyedrop raises aqueous humor levels within a period of 30–60 min and mostly the levels fall within 4 h. Being underprivileged organ, eye lacks first-line immune defense mechanisms in the humors, antimicrobial drug levels above the MIC of microbe is required for their activity. Therefore, during corneal infections, hourly or 2 hourly instillations of antimicrobial agents are preferred. To estimate the appropriate drug schedule, pharmacokinetic simulations are helpful. Aqueous humor kinetics of single and multiple doses of topical, non-preserved voriconazole (VZ) were studied in human eyes comparing hourly versus 2 hourly instillations (Senthilkumari et al. 2010). Single-dose ocular kinetics of 1 % VZ resulted in a maximum mean aqueous concentration of 3.333 ± 1.61 μg/ml in 30 min; multidose kinetic study revealed that hourly and bihourly dosing resulted in mean aqueous concentrations of 7.47 ± 2.14 μg/ml and 4.69 ± 2.7 μg/ml, respectively (Fig. 3.3).
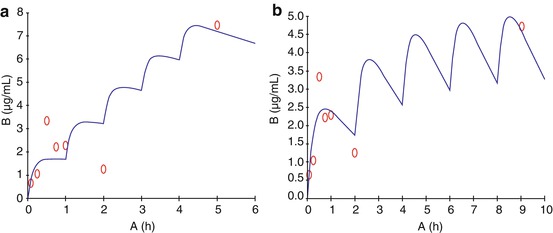
Fig. 3.3
PK simulation from our laboratory showing the predicted concentration of voriconazole in aqueous humor achieved hourly (A), 2 hourly (5 doses) instillation of voriconazole topical application in patients undergoing cataract surgery (o observed, — predicted) (Senthilkumari et al. 2010)
Serial sampling of the humors of the eye is not a feasible task in human or animals while conducting ocular pharmacokinetic studies. Therefore, the model of collecting samples at various time points after the administration of drug is followed in all the studies except few studies indicating the worth of microdialysis for serial sampling in animals. Many of the ocular drugs were investigated in the literature for their intraocular penetration, but only few of them were interpreted with the help of pharmacokinetic models to explain their ocular disposition.
After a single topical instillation of tritiated clonidine HCl solution (0.2 %) at the volume of 30 µl into rabbit eyes, Chiang and Schoenwald (1986) evaluated clonidine ocular pharmacokinetics. Seven different tissues and plasma were excised and assayed for drug concentration over 180 min. They have also generated data at steady state levels of drug in cornea using an infusion assembly. The data were analysed by fitting them into physiological and a classical diffusion pharmacokinetic models. This study reported that the physiological model parameters were fit to the topical infusion data and showed good agreement between the predicted and experimental data.
3.2.4 Drug Interaction in Topically Applied Drugs
More than one eyedrops are often required to be administered together at times. In such conditions, chemical incompatibility has been observed which is causing one of them to precipitate in the lower fornix. This is a potential cause for the reduction of pre-corneal drug availability of the topically applied drugs. Ciprofloxacin hydrochloride deposition on the lower fornix of the eye due to its pH incompatibility has been claimed as the advantage of pre-corneal deposits as drug depots “ciprofloxacin HCl precipitates.” However, redistribution or dissolution of these deposits back to increase tear film concentrations have not been proved. Ocular pharmacokinetics of the topically applied drug also gets altered by coadministered drugs when they are competing for the same transport mechanism (Nirmal et al. 2013a). Coadministration of local anesthetics and antimuscarinic agents might increase ocular bioavailability of the drugs by decreasing tear film secretion. This interaction so far is attributed to the decrease in tear film secretion; however, other interactions on the corneal transport mechanism require further studies.
3.2.5 Ocular Drug Levels after Systemic Administration
Ocular pharmacokinetics of 1,3-bis(2-chloroethyl)-1-nitrosourea (BCNU) was investigated using its levels in vitreous and aqueous humors after its systemic administration in rabbits. This study compared systemic, topical, and subconjunctival administrations and suggested that topical application of BCNU is the best and subconjunctival injection the second best route for treating an iris tumor and that intravenous injection is the best route for choroidal and retinal tumors (Ueno et al. 1982).
Aqueous humor penetration of amikacin was assessed in the anterior chamber of the human eyes using radioimmunoassay. This study reported that bactericidal concentrations of amikacin were not achieved by topical or intravenous administration. Subconjunctival injection also did not produce consistent bactericidal concentration of amikacin in the aqueous humor. This study has also reported that iris pigment binding as one of the factor responsible for the poor levels reached (Eiferman and Stagner 1982). Several clinical studies showed that inadequate penetration of antimicrobial agents into the eye after their systemic administration as a limitation for ocular infections (Velpandian 2009). Only after multiple doses orally administered fluoroquinolones like ciprofloxacin reached significant levels in the vitreous above the MIC for most of the ocular pathogenic organisms (Keren et al. 1991). Inadequate penetration of different classes of drugs into non-inflammed eyes after systemic administration has been well documented by many investigations.
3.2.6 Factors Affecting Ocular Penetration of Drugs
Aqueous ophthalmic drug solutions typically exhibit low bioavailability due to various loss processes such as drainage, tear turnover, nonproductive absorption, and protein binding. Suspensions may improve bioavailability, but because of a short residence time and a low corneal permeability rate constant, the dissolution rate of the drug and its intrinsic solubility must be considered.
Topically applied hydrophobic compounds like amphotericin B showed undetectable levels of drug after single application; however, after repeated application, therapeutic levels reached the cornea in the normal eyes of rabbits. In inflamed eyes the levels reached high quickly but fell rapidly (O’Day et al. 1986). Corneal inflammation that induced increased drug penetration across the cornea is not well documented in the literature due to the difficulties encountered in sampling aqueous humor from patients undergoing ocular surgeries.
3.2.7 Ocular Drug Metabolism
Although the liver is still the major organ involved in the biotransformation of drugs, drug metabolism in the eye is often pursued as a avenue for the development of designer drugs. The corneal epithelium is the source of amidases and esterases which favors the optimization of dosage forms using the concept of prodrugs. Latanoprost and dipivefrin are the best examples for the prodrugs to get into their active form after the metabolism in the cornea. In the cornea, the presence of aminopeptidases and other peptidases has also been reported, and their involvements in the metabolism of drugs are confirmed (Lee et al. 1986). While studying the ocular pharmacokinetics of topically applied phenylephrine, Antoine et al. (1984) reported that the corneal epithelium is responsible for the metabolic degradation of phenylephrine which occurred following its topical instillation. While working with levobunolol for topical antiglaucoma treatment, Tang-Liu and coworkers (1987) demonstrated that the major sites of ocular metabolism were the corneal epithelium and the iris-ciliary body. On passage across the cornea, 4.7 % of topically applied levobunolol dose was biotransformed to an active metabolite dihydrolevobunolol and subsequently became bioavailable to intraocular tissues. Another 12 % of the topical levobunolol dose entered the systemic circulation as metabolite after pre-systemic biotransformation. Nakamura et al. (2005) examined the expression levels of the different conjugation enzymes, sulfotransferases, UDP-glucuronosyl transferases (UGTs), and glutathione S-transferases (GSTs), in ocular tissues. In 5-week-old animals, the CYP genes, CYP2B2 and CYP3A1, were abundantly expressed in the lens, with higher CYP1A1 expression detectable in the extra-lenticular tissues, of both genders. They have also reported that in general, the expression levels of the CYPs and sulfotransferases declined with age, whereas the levels of the UGTs and GSTs increased. These results demonstrate that the expression profiles of drug-metabolizing enymes show both region- and age-specific patterns in rat ocular tissues. Presence of various metabolizing enzymes and their physiological function has been targeted for ocular drug delivery (Duvvuri et al. 2004).
3.2.8 Elimination Pathways of the Drugs
A major pathway for systemic absorption of topically applied drug has been reported to be through the walls of the gastrointestinal tract (Anderson 1980). Schmitt et al. (1980) evaluated the penetration of radiolabeled timolol into the rabbit eye after topical instillation and after intravenous injection in animals. This study reported that the levels of radioactivity were considerably greater in ocular tissues after instillation as compared with intravenous injection, whereas in extraocular tissues, the levels were similar after both routes of administration. This study has also reported that in ocular instillation, only unchanged timolol was present in the aqueous humor, whereas both timolol and metabolites were present in the serum. After intravenous administration, timolol was rapidly metabolized and metabolites appeared in the serum and aqueous humor. Pharmacokinetic studies conducted after the intravitreal injection of drugs revealed that two predominating pathways are involved in the clearance of drugs. Most of the drugs are cleared through posterior elimination pathway through retinal blood vessels Drugs like aminoglycoside antibiotics follow anterior pathway through aqueous humor and iris (Barza et al. 1983; Gupta et al. 2000; Nirmal et al. 2012).
3.2.9 Functional Importance of Blood-Ocular Barriers Affecting Pharmacokinetics
Drug penetration across blood-ocular barriers is now well recognized; therefore, beyond pharmaceutical parameters, their susceptibility for various transporters is expected to govern their pharmacokinetics. Probenecid (Benemid) was the agent first explored to elevate serum penicillin concentrations (Burnell and Kirby 1951). The interest to evaluate the impact on the concentration of penicillin derivatives in ocular fluids (Barza et al. 1973; Salminen 1978). However, these studies showed that administration of probenecid had an enhancing effect on ocular cloxacillin concentration allowing improved drug diffusion into the eye by means of an elevated plasma concentration and had no specific ocular effect. In the subsequent studies after the intravitreal administration of carbenicillin, concomitant intraperitoneal dosing of probenecid prolonged the vitreal half-life of carbenicillin and showed that beta-lactam antibiotics are eliminated via the retinal route. Ever since, observations and curiosities among the researchers led to better fundamental understanding regarding the involvement of drug transporters in blood-ocular barriers. Using retinal capillary endothelial cell lines presence and function of various transporters were explored (Hosoya and Tomi 2005; Mannermaa et al. 2006). Subsequently, functional importance of such transporters for the penetration of xenobiotics reported in animals (Senthilkumari et al. 2009; Nirmal et al 2012; Gunda et al. 2006)
Although, the presence of transporter proteins in the blood-ocular barriers has been well documented, their extent and role affecting pharmacological action have come to light after the systematic explorations in the last decade. In these controlled experiments, probes were used as a substrate and their pharmacokinetic modulation was assessed after blocking the functions of transporters with respective agents.
Functional role of P-gp and ocular tissue distribution of intravitreally injected rhodamine 123 (Rho-123) was evaluated in the presence of P-gp-specific blocker (GF 120918) in normal as well as rifampicin-fed rabbits using microdialysis and direct sampling technique. This study revealed that intravenously injected blocker significantly altered the ocular disposition of intravitreally injected P-gp substrate. Rifampicin pretreatment did not upregulate P-gp transporters of the retina to the extent to affect the intravitreal kinetics of Rho-123 significantly (Senthilkumari et al. 2008a). The impact of P-glycoprotein (P-gp) blockade on the intravenous (i.v.) pharmacokinetics of Rho-123 and the subsequent effect on its disposition in ocular and non-ocular tissues were studied using rabbits. This study concluded that increasing the ocular concentration of systemically given drugs may not be possible with the degree of P-gp blockade achieved when using GF120918 (P-gp blocker) at the studied concentration after intravenous administration (Senthilkumari et al. 2008b). The effect of P-glycoprotein modulation at blood-ocular barriers using gamma scintigraphy in rabbits is shown in Fig. 3.4. This confirmed the involvement of P-glycoprotein in the intraocular disposition of susceptible drugs (Senthilkumari et al. 2009).
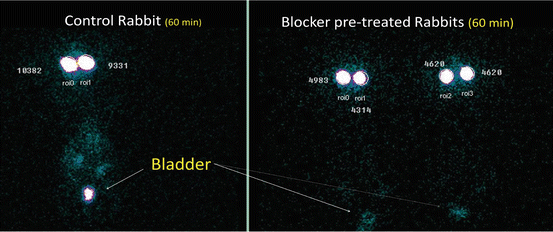
Fig. 3.4
Static planar gamma camera image of rabbits following intravitreal injection of 99mTc-ofloxacin in rabbits. Note: Control rabbit bladder showing higher radioactivity
Although aminoglycoside antibiotics were reported to be cleared through the anterior route (Barza et al. 1983), while studying OCT transporters, Nirmal et al. (2013b) concluded that the clearance of organic cation transporter (OCT) substrates favors the anterior elimination pathway. The functional importance of (OCT) on the ocular disposition of intravitreally injected substrate tetraethyl ammonium (TEA) was assessed in rabbits (Nirmal et al. 2013b); this study concluded that intravitreally injected OCT substrates may follow an anterior elimination pathway and prolonged residence time in the vitreous humor. This study showed that OCT may not be active from vitreous to blood route in the blood-retinal barrier.
The potential pharmacokinetic role of organic cation transporters in modulating the trans-corneal penetration of its substrates administered topically has been studied in rabbits. This study concluded that OCT is functionally active in the cornea causing uptake of their substrates from tear to the aqueous humor. When administering their substrates/blockers topically, both may be competing for OCT for their uptake across the cornea, thereby decreasing the corneal penetration. Hence, OCT can have a potential pharmacokinetic role in modulating the ocular bioavailability of their substrates which are used topically for ocular therapeutics (Nirmal et al. 2013b). The role of organic cation transporters was studied in the ocular disposition of its intravenously injected substrate in rabbits by quantifying the levels of its substrate in the presence and absence of blockers. This study revealed that in most of the tissues, OCTs are functionally present from apical to basolateral. The gene expression studies also showed the presence of OCT1, OCTN1, and OCTN2 in various ocular tissues studied. This study suggested that OCTs are functionally active in blood-ocular barriers and involved in the transport of its substrate from blood to vitreous humor (Nirmal et al. 2013a). Moreover, this study also revealed the pre-corneal availability of OCT substrate through lacrimal secretion indicating the possibility of utilizing them for the delivery of drug to the tear film through systemic route (Nirmal et al. 2010). Apart from OCT, OAT, and P-gp transporters, PEPT transporters have been used for the delivery of stable dipeptide prodrugs for improved absorption of acyclovir (Talluri et al. 2008).
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


