13
Drug Therapy of Neurodegenerative Diseases
This chapter will be most useful after having a basic understanding of the material in Chapter 22, Treatment of Central Nervous System Degenerative Disorders in Goodman & Gilman’s The Pharmacological Basis of Therapeutics, 12th Edition. In addition to the material presented here, the 12th Edition includes:
• A discussion of the selective vulnerabilities of specific CNS neurons
• A detailed discussion of the roll of genetics and environment in neurodegenerative disorders
• The common cellular mechanisms of neurodegeneration
• A detailed discussion of the neural mechanisms and neuroprotective mechanisms of parkinsonism
• A complete discussion of the genetics of Huntington’s disease
• Chemical structures of the drugs used to treat and manage neurodegenerative disorders
• Figure 22-8 Monosynaptic muscle stretch reflex with descending control via inhibitory interneurons
LEARNING OBJECTIVES
 Understand the pathophysiology of neurodegenerative diseases, including Parkinson’s disease (PD), Alzheimer’s disease (AD), Huntington’s disease (HD), and amyotrophic lateral sclerosis (ALS).
Understand the pathophysiology of neurodegenerative diseases, including Parkinson’s disease (PD), Alzheimer’s disease (AD), Huntington’s disease (HD), and amyotrophic lateral sclerosis (ALS).
 Understand the role of drugs in the treatment and management of the symptoms of neurodegenerative diseases.
Understand the role of drugs in the treatment and management of the symptoms of neurodegenerative diseases.
 Know the mechanisms of action and the adverse effects of drugs that are used to treat and manage neurodegenerative diseases.
Know the mechanisms of action and the adverse effects of drugs that are used to treat and manage neurodegenerative diseases.
DRUGS INCLUDED IN THIS CHAPTER
• Amantadine (SYMMETREL)
• Apomorphine (APOKYN)
• Baclofen (LIORESAL)
• Carbidopa (LODOSYN)
• Carbidopa/levodopa (SINEMET, ATAMET, others; orally disintegrating tablet, PARCOPA)
• Dantrolene (DANTRIUM)
• Donepezil (ARICEPT)
• Entacapone (COMTAN; fixed combination with carbidopa/levodopa, STAVELO)
• Galantamine (NIVALIN, others)
• Levodopa (L-DOPA, LARODOPA)
• Memantine (NAMENDA)
• Pramipexole (MIRAPEX)
• Rasagiline (AZILECT)
• Riluzole (RILUTEK)
• Rivastigmine (EXELON)
• Ropinirole (REQUIP)
• Selegiline (ELDEPRYL; oral disintegrating tablet, EMSAM; transdermal patch, ZELAPAR)
• Tacrine (COGNEX)—discontinued in the United States
• Tetrabenazine (XENAZINENITOMAN)
• Tizanidine (ZANAFLEX)
• Tolcapone (TASMAR)
MECHANISMS OF ACTION OF DRUGS USED TO TREAT AND MANAGE NEURODENERATIVE DISORDERS
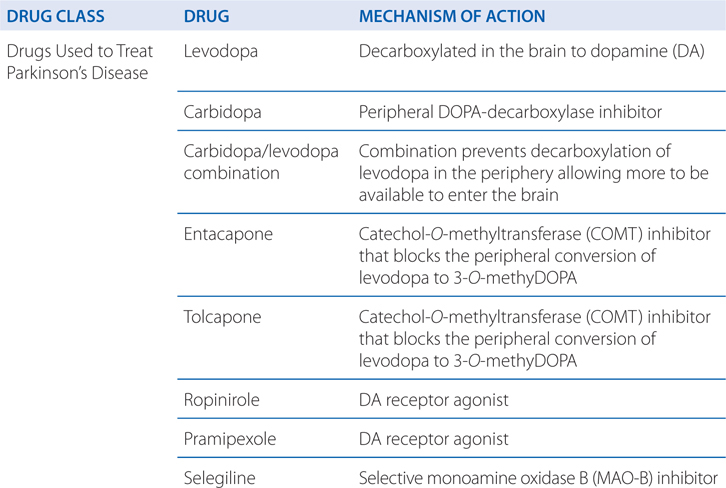
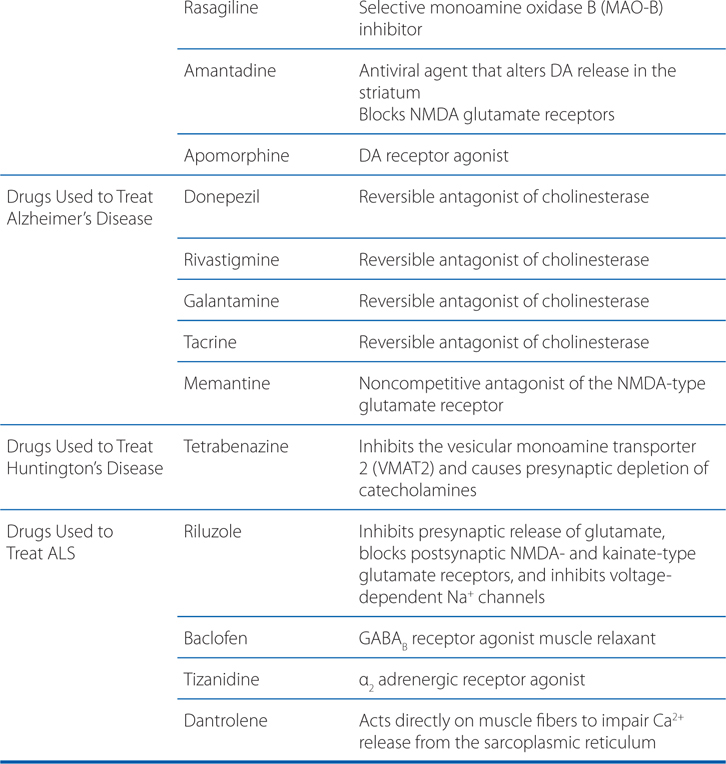
A 68-year-old man is diagnosed with Parkinson’s disease (PD).
a. What is the pathophysiology of Parkinson’s disease?
The dopaminergic deficit in PD arises from a loss of the neurons in the substantia nigra pars compacta that provide innervation to the striatum (caudate and putamen). The current understanding of the pathophysiology of PD is based on the finding that the striatal DA content is reduced in excess of 80%. This paralleled the loss of neurons from the substantia nigra, suggesting that replacement of DA could restore function.
b. How is dopamine synthesized in neurons?
DA, a catecholamine, is synthesized in the terminals of dopaminergic neurons from tyrosine and stored, released, and metabolized by processes described in Chapter 5 and summarized in Figure 13-1. The actions of DA in the brain are mediated by a family of DA receptor proteins. Two types of DA receptors are identified in the mammalian brain using pharmacological techniques: D1 receptors, which stimulate the synthesis of the intracellular second messenger cyclic AMP; and D2 receptors, which inhibit cyclic AMP synthesis as well as suppress Ca2+ currents and activate receptor-operated K+ currents.
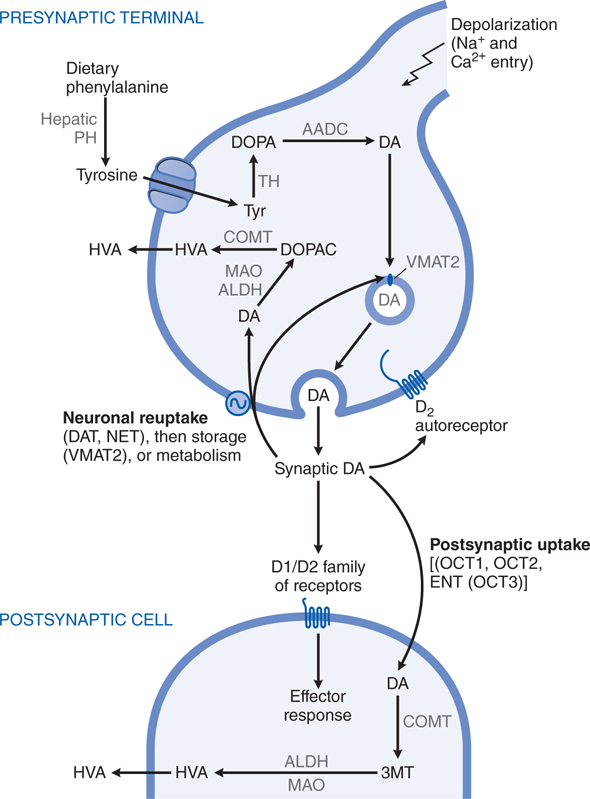
FIGURE 13-1 Dopaminergic nerve terminal. Dopamine (DA) is synthesized from tyrosine in the nerve terminal by the sequential actions of tyrosine hydrolase (TH) and aromatic amino acid decarboxylase (AADC). DA is sequestered by VMAT2 in storage granules and released by exocytosis. Synaptic DA activates presynaptic autoreceptors and postsynaptic D1 and D2 receptors. Synaptic DA may be taken up into the neuron via the DA and NE transporters (DAT, NET), or removed by postsynaptic uptake via OCT3 transporters. Cytosolic DA is subject to degradation by monoamine oxidase (MAO) and aldehyde dehydrogenase (ALDH) in the neuron, and by catechol-O-methyltransferase (COMT) and MAO/ALDH in non-neuronal cells; the final metabolic product is homovanillic acid (HVA). See structures in Figure 13-4. PH, phenylalanine hydroxylase.
CARDINAL FEATURES OF PARKINSON DISEASE (PD)
• Bradykinesia (slowness and poverty of movement)
• Muscular rigidity
• Resting tremor (which usually abates during voluntary movement)
• Impairment of postural balance leading to disturbances of gait and falling
c. How is dopamine neurotransmission affected by Parkinson’s disease?
Considerable effort has been devoted to understanding how the loss of dopaminergic input to the neurons of the neostriatum gives rise to the clinical features of PD. The basal ganglia can be viewed as a modulatory side loop that regulates the flow of information from the cerebral cortex to the motor neurons of the spinal cord (see Figure 13-2). The neostriatum is the principal input structure of the basal ganglia and receives excitatory glutamatergic input from many areas of the cortex. Most neurons within the striatum are projection neurons that innervate other basal ganglia structures. A small but important subgroup of striatal neurons consists of interneurons that connect neurons within the striatum but do not project beyond its borders. Acetylcholine (ACh) and neuropeptides are used as transmitters by these striatal interneurons.
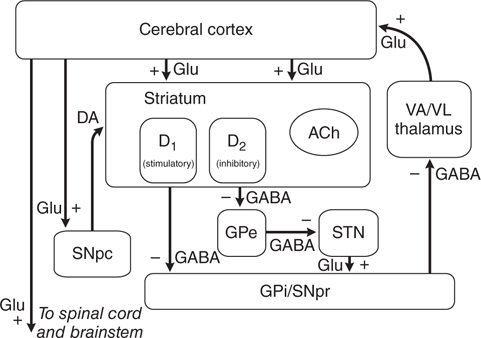
FIGURE 13-2 Schematic wiring diagram of the basal ganglia. The striatum is the principal input structure of the basalganglia and receives excitatory glutamatergic input from many areas of cerebral cortex. The striatum contains projection neurons expressing predominantly D1 or D2 dopamine receptors, as well as interneurons that use acetylcholine (ACh) as a neurotransmitter. Outflow from the striatum proceeds along two routes. The direct pathway, from the striatum to the substantianigra pars reticulata (SNpr) and globus pallidus interna (GPi), uses the inhibitory transmitter GABA. The indirect pathway, from the striatum through the globus pallidus externa (GPe) and the subthalamic nucleus (STN) to the SNpr and GPi consists of two inhibitory GABAergic links and one excitatory glutamatergic projection (Glu). The substantia nigra pars compacta (SNpc) provides dopaminergic innervation to the striatal neurons, giving rise to both the direct and indirect pathways, and regulates the relative activity of these two paths. The SNpr and GPi are the output structures of the basal ganglia and provide feedback to the cerebral cortex through the ventroanterior and ventrolateralnuclei of the thalamus (VA/VL).
The key feature of this model of basal ganglia function, which accounts for the symptoms observed in PD as a result of loss of dopaminergic neurons, is the differential effect of DA on the direct and indirect pathways (see Figure 13-3). The dopaminergic neurons of the substantia nigra pars compacta (SNpc) innervate all parts of the striatum; however, the target striatal neurons express distinct types of DA receptors. The striatal neurons giving rise to the direct pathway express primarily the excitatory D1 dopamine receptor protein, whereas the striatal neurons forming the indirect pathway express primarily the inhibitory D2 type. Thus, DA released in the striatum tends to increase the activity of the direct pathway and reduce the activity of the indirect pathway, whereas the depletion that occurs in PD has the opposite effect. The net effect of the reduced dopaminergic input in PD is to increase markedly the inhibitory outflow from the SNpc and globus pallidus interna (GPi) to the thalamus and reduce excitation of the motor cortex.
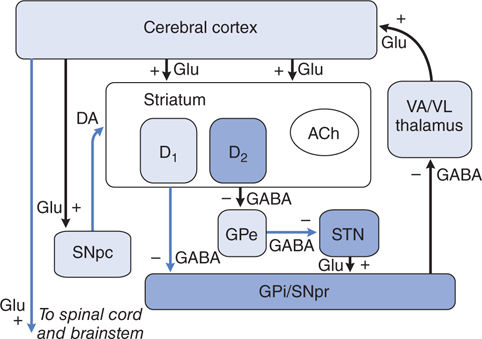
FIGURE 13-3 The basal ganglia in Parkinson’s disease. The primary defect is destruction of the dopaminergic neurons of the SNpc. The striatal neurons that form the direct pathway from the striatum to the SNpr and GPi express primarily the excitatory D1 dopamine receptor, whereas the striatal neurons that project to the GPe and form the indirect pathway express the inhibitory D2 dopamine receptor. Thus, loss of the dopaminergic input to the striatum has a differential effect on the two outflow pathways; the direct pathway to the SNpr and GPi is less active (structures in light blue), whereas the activity in the indirect pathway is increased (structures in dark blue). The net effect is that neurons in the SNpr and GPi become more active. This leads to increased inhibition of the VA/VL thalamus and reduced excitatory input to the cortex. (See legend to Figure 13–2 for definitions of anatomical abbreviations.)
d. What is the prognosis for this patient?
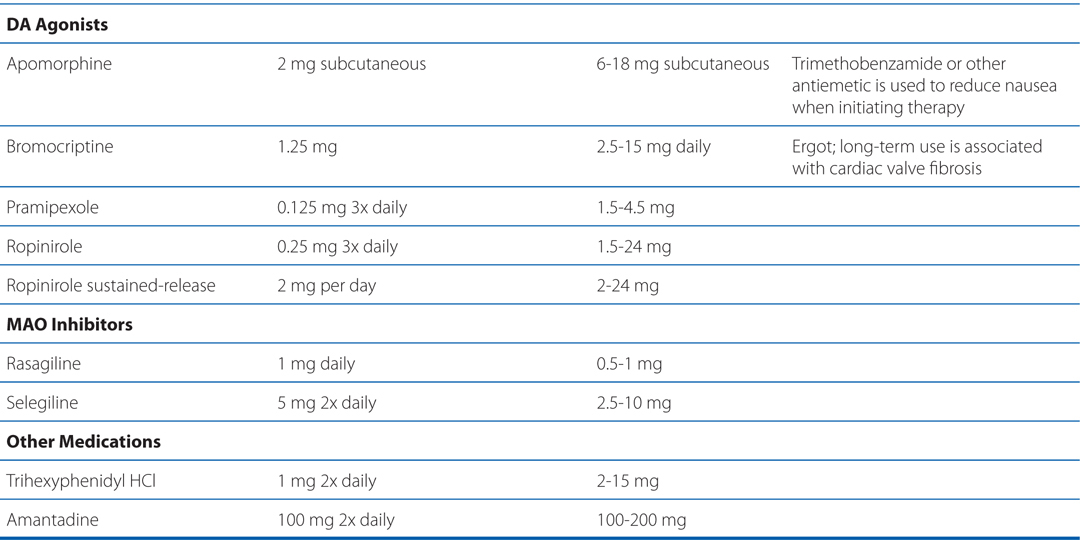
Progressive loss of dopamine (DA)-containing neurons is a feature of normal aging; however, most people do not lose the 70 to 80% of dopaminergic neurons required to cause symptomatic PD. Without treatment, PD progresses over 5 to 10 years to a rigid, akinetic state in which patients are incapable of caring for themselves. Death frequently results from complications of immobility, including aspiration pneumonia or pulmonary embolism. The availability of effective pharmacological treatment has radically altered the prognosis of PD; in most cases, good functional mobility can be maintained for many years. Life expectancy of adequately treated patients is increased substantially, but overall mortality remains higher than that of the general population. Commonly used medications for the treatment of PD are summarized in Table 13-1.
TABLE 13-1 Commonly Used Medications for the Treatment of Parkinson’s Disease

The patient in Case 13-1 is early in the course of his Parkinson’s disease
a. What are the treatment options at this stage of his disease?
Commonly used medications for the treatment of PD are summarized in Table 13-1. Pharmacological treatment of PD should be tailored to the individual patient. Drug therapy is not obligatory in early PD; many patients can be managed for a time with exercise and lifestyle interventions. For patients with mild symptoms, MAO-B inhibitors, amantadine, or (in younger patients) anticholinergics are reasonable choices. In most patients, treatment with a dopaminergic drug, either levodopa or a DA agonist, is eventually required. Large controlled clinical trials provide convincing evidence for a reduced rate of motor fluctuation in patients in which DA agonists are used as initial treatment. This benefit was, however, accompanied by an increased rate of adverse effects, especially somnolence and hallucinations. Practitioners prefer a DA agonist as initial therapy in younger patients in order to reduce the occurrence of motor complications. In older patients or those with substantial comorbidity, levodopa/carbidopa is generally better tolerated.
As this (the patient from Case 13-1) patient’s disease progresses, it is recommended that he be treated with levodopa.
a. What is levodopa, and how is it used in the treatment of Parkinson’s disease?
Levodopa, the metabolic precursor of DA, is the single most effective agent in the treatment of PD. Levodopa is itself largely inert; both its therapeutic and adverse effects result from the decarboxylation of levodopa to DA. The metabolism of levodopa is shown in Figure 13-4.
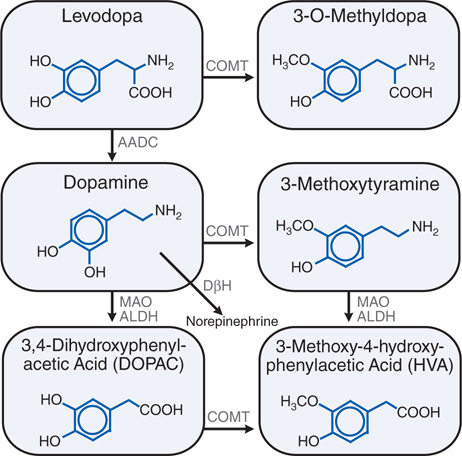
FIGURE 13-4 Metabolism of levodopa (L-DOPA). ALDH, aldehyde dehydrogenase; COMT, catechol-O-methyltransferase; DβH, dopamine β-hydroxylase; AADC, aromatic L-amino acid decarboxylase; MAO, monoamine oxidase.
Entry of levodopa into the CNS across the blood-brain barrier is mediated by a membrane transporter for aromatic amino acids, and competition between dietary protein and levodopa may occur at this level. In the brain, levodopa is converted to DA by decarboxylation primarily within the presynaptic terminals of dopaminergic neurons in the stratium (see Figure 13-5). The DA produced in the presynaptic terminals is responsible for the therapeutic effectiveness of levodopa in PD; after release, it is either transported back into dopaminergic terminals by the presynaptic uptake mechanism or metabolized by the actions of MAO and catechol-O-methyltransferase (COMT) (see Figure 13-4).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


