KEY CONCEPTS
![]() Chronic kidney disease (CKD) results in minimal alterations in the absorption or bioavailability of most drugs.
Chronic kidney disease (CKD) results in minimal alterations in the absorption or bioavailability of most drugs.
![]() The volume of distribution (VD) of many drugs is increased in the presence of acute and chronic kidney disease as a consequence of volume expansion and/or reduced protein binding.
The volume of distribution (VD) of many drugs is increased in the presence of acute and chronic kidney disease as a consequence of volume expansion and/or reduced protein binding.
![]() In addition to the expected decrement in renal clearance, nonrenal clearance (i.e., GI and hepatic drug metabolism) of several drugs is also reduced in patients with CKD.
In addition to the expected decrement in renal clearance, nonrenal clearance (i.e., GI and hepatic drug metabolism) of several drugs is also reduced in patients with CKD.
![]() Individualization of a drug dosage regimen for a patient with reduced kidney function is based on the pharmacodynamic/pharmacokinetic characteristics of the drug and the patient’s degree of residual renal function.
Individualization of a drug dosage regimen for a patient with reduced kidney function is based on the pharmacodynamic/pharmacokinetic characteristics of the drug and the patient’s degree of residual renal function.
![]() The drug dosing guidelines for patients with CKD in many drug information resources are highly variable and many are not optimal for clinical use.
The drug dosing guidelines for patients with CKD in many drug information resources are highly variable and many are not optimal for clinical use.
![]() The effect of hemodialysis or peritoneal dialysis on drug elimination is dependent on the characteristics of the drug and the dialysis prescription.
The effect of hemodialysis or peritoneal dialysis on drug elimination is dependent on the characteristics of the drug and the dialysis prescription.
![]() Hemodialysis clearance data can be used to guide the initial drug dosage regimen recommendation for hemodialysis patients; however, prospective monitoring of serum concentrations is often warranted especially for those with a narrow therapeutic index.
Hemodialysis clearance data can be used to guide the initial drug dosage regimen recommendation for hemodialysis patients; however, prospective monitoring of serum concentrations is often warranted especially for those with a narrow therapeutic index.
Chronic kidney disease (CKD) is a common condition characterized by the presence of kidney damage, a urine albumin-to-creatinine ratio greater than 30 mg/g (3.4 mg/mmol) or a glomerular filtration rate (GFR) of less than 60 mL/min (1 mL/s) for greater than 3 months. It is estimated that 10% of adults, greater than 20 million people, in the United States have CKD.1 Between 2000 and 2008, the incidence of CKD has more than doubled in adults 65 years old and older.1 The combination of CKD, age-related reductions in renal function, and the high utilization of medications in these patients increases their risk of adverse effects particularly if their drug regimen is not appropriately individualized.2,3 Furthermore a marked reduction in kidney function in adults of any age, whether acute or chronic, has been noted to affect the pharmacokinetics of many medications.4–12 Thus patients with CKD are often prescribed an extensive array of medications for CKD-related and other comorbid conditions (see Chap. 29). These factors necessitate that clinicians individualize drug therapy to maximize therapeutic outcomes and to minimize therapeutic misadventures.
Drug excretion is markedly effected by CKD. Medications which are predominantly renally eliminated unchanged (fe) may accumulate in patients with CKD, which can complicate existing conditions and increase the risk of adverse effects. If 30% or more of a drug is renally eliminated unchanged, it will have a high likelihood, >70%, of requiring dosage regimen adjustment in patients with CKD. The pharmacokinetics of drugs with an fe less than 30% also may be affected and a recent study indicated that 20% to 37.5% of such drugs approved in the United States from 1998 to 2010 had a dosage adjustment for patients in the product labeling.13 If there is no official recommendation in the product labeling a dosage regimen adjustment may be calculated on the basis of the drug’s fe and the ratio of the patient’s residual renal function relative to an age and gender normal value for estimated creatinine clearance (CLcr) or GFR.14 However, for medications that are extensively metabolized or for which dramatic changes in protein binding and/or distribution volume (VD) have been noted, a more complex adjustment strategy may need to be employed.4,15 Furthermore, physiologic and biochemical changes associated with CKD may also independently impact drug dosing and serum drug concentrations.4
Clinicians thus will often need to proactively design individualized therapeutic regimens to optimize achievement of the desired outcomes while minimizing adverse events, if they use basic pharmacokinetic principles combined with the drug’s disposition characteristic and the patient’s clinical status. In this chapter, the influence of CKD on drug pharmacokinetic properties is characterized and the most commonly used medications that are affected are discussed. In addition, a general guide for individualizing drug therapy in patients with CKD is presented along with dosage recommendations for the most commonly used drugs in this patient population. Finally, the impact of chronic renal replacement therapy (i.e., peritoneal dialysis and hemodialysis) on drug disposition is discussed and dosage recommendations for selected drugs are presented. Drug dosage regimen adjustment strategies for patients with acute kidney injury (AKI) including those who are receiving continuous renal replacement therapy are presented in Chapter 28.
EFFECT OF CHRONIC KIDNEY DISEASE ON DRUG DISPOSITION
Prior to the 1990s, there were no regulatory guidelines regarding when and how pharmacokinetic/pharmacodynamic studies of drugs in patients with renal insufficiency should be conducted as part of the new drug application process.16,17 Thus much of the pharmacokinetic/pharmacodynamic data in patients with renal insufficiency published during the 1970s through much of the 1990s were derived from postmarketing studies. Unfortunately, the utility of these studies were often limited by small sample sizes, which led to inconsistent and sometimes conflicting results. Furthermore it was rare that detailed information was incorporated into the official (i.e., FDA) prescribing information. The FDA published the first guidance for industry on renal impairment studies in 1998 and since then, the frequency and quality of renal impairment studies as part of drug development has increased.13,18,19 A proposed revision to the 1998 guidance on renal impairment studies was recently circulated for comment which recommended: (a) conducting studies in nonrenally as well as renally eliminated drugs, (b) conducting studies in patients receiving hemodialysis, (c) conducting studies to evaluate pharmacokinetics of therapeutic proteins in patients with renal insufficiency, (d) categorizing renal function based on estimated GFR (using the modification of diet in renal disease [MDRD] equation) or CLcr (using the Cockcroft–Gault [C–G] equation), and (e) modifications to how the results of renal impairment studies are presented in the official drug label.20–22 Although this revision has not been “officially” implemented by the FDA, it appears to be impacting how the industry considers the design of renal pharmacokinetic/pharmacodynamic studies.
Drug Absorption
![]() There is little quantitative information regarding the influence of CKD on drug absorption and bioavailability. Many studies evaluating bioavailability of oral medications in CKD patients were not designed to provide an assessment of the drug’s absolute bioavailability (e.g., they did not include a comparison of the area under the concentration–time curve (AUC) after oral and IV administration of the drug). Rather, the principal outcomes that were documented were alterations in the peak concentration (Cmax), time at which the peak concentration was attained (tmax), or in the fractional amount of drug recovered in the urine in a finite time period. Unfortunately, this limited information has been extrapolated by some into a general conclusion that drug absorption is slowed and/or that the extent of absorption is reduced in patients with CKD.23,24
There is little quantitative information regarding the influence of CKD on drug absorption and bioavailability. Many studies evaluating bioavailability of oral medications in CKD patients were not designed to provide an assessment of the drug’s absolute bioavailability (e.g., they did not include a comparison of the area under the concentration–time curve (AUC) after oral and IV administration of the drug). Rather, the principal outcomes that were documented were alterations in the peak concentration (Cmax), time at which the peak concentration was attained (tmax), or in the fractional amount of drug recovered in the urine in a finite time period. Unfortunately, this limited information has been extrapolated by some into a general conclusion that drug absorption is slowed and/or that the extent of absorption is reduced in patients with CKD.23,24
Both CKD and comorbid conditions associated with CKD (e.g., diabetes mellitus and cardiovascular disease) may contribute to changes in drug absorption. Some patients present with changes in GI transit time and gastric pH (e.g., gastroparesis associated with diabetes mellitus), edema of the GI tract, persistent vomiting and diarrhea, and phosphate binder administration have all been proposed as a rationale for alterations in the bioavailability of drugs in CKD patients.2,8 For example, patients with uremic-induced vomiting, now only occasionally observed in patients with severe renal insufficiency, may decrease a medication’s time in the GI tract and thereby limit absorption of that drug. Edema of the GI tract, secondary to cirrhosis or congestive heart failure, can decrease the bioavailability of some medications such as oral furosemide.2,25 A reduction in gastric acidity associated with the concomitant administration of antacids and phosphate binders has been associated with a reduction in bioavailability of several medications including several antibiotics and digoxin.26–29
However, there are only a few drugs (e.g., some β-blockers, dextropropoxyphene, dihydrocodeine, felodipine, sertraline, and cyclosporine) with documented increases in bioavailability in patients with CKD as a result of a reduction in metabolism during the drug’s first pass through the GI tract and liver.4,30–35 Although the bioavailability of several of these compounds is increased in the presence of CKD, clinical consequences (development of excessive or unexpected adverse effects) have only been demonstrated with dextropropoxyphene and dihydrocodeine.31,32 For some agents, those metabolized by cytochrome P450 (CYP) 3A4, the bioavailability may be dramatically increased when the medications are taken along with grapefruit juice: felodipine by 184% and cyclosporine by 20%.33,34
Drug Distribution
The VD of many drugs is increased in patients with moderate to severe CKD as well as those with preexisting CKD who develop AKI (Table 33-1) and lead to a reduction in serum drug concentrations.4,8–10,36 This increase in VD may be the result of pathophysiologic alterations in body composition, fluid overload secondary to excessive IV fluid administration, decreased protein binding, or increased tissue binding. Decreased tissue binding of drugs in patients with CKD may result in a reduction in VD, which has been reported for only a few medications (e.g., digoxin and pindolol).9,10
TABLE 33-1 Volume of Distribution of Selected Drugs in Patients with End-Stage Renal Disease
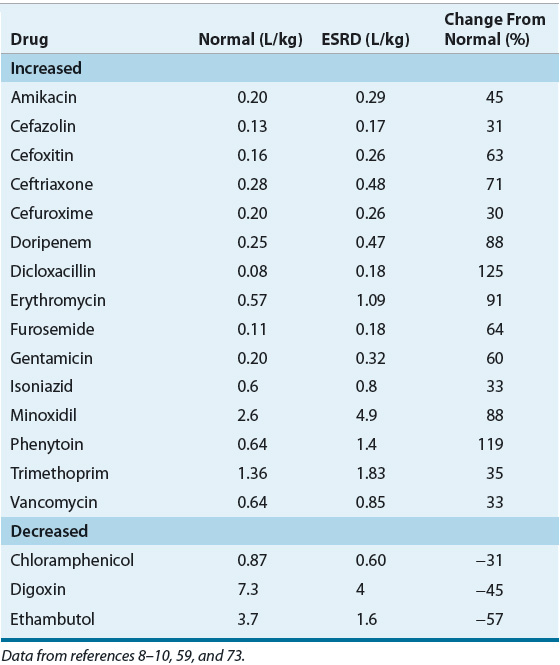
Variability in fluid status is a common issue in patients with CKD, especially those that are critically ill. Many of these critically ill patients receive large volumes of IV fluids for various clinical conditions (e.g., resuscitation from shock), and develop edema, pleural effusions, or ascites as a result thereof. These therapeutic interventions, in addition to renal insufficiency that can result in reduced water excretion, can result in an increase in VD and alterations in drug serum concentrations, especially with hydrophilic drugs.3 For example the VD of aminoglycosides has been noted to be increased by 1.34- to 1.66-fold in postsurgical patients with septic shock.37 Interstitial fluid concentration of piperacillin was also reported to be three- to fourfold lower in patients undergoing heart surgery and in patients with sepsis.37 These marked reductions in drug concentration may be due to increased capillary permeability following fluid accumulation in the interstitial space.37
Effect of Altered Protein Binding
![]() The unbound fraction of many acidic drugs increases in CKD patients as the result of a decrease in protein binding and this is associated with an increase in the apparent VD.8 A new equilibrium is ultimately established as a result of increased drug elimination/distribution, such that the unbound concentrations remain comparable to those observed in patients with normal renal function despite the fact that total concentrations are reduced. Thus, the net effect is an alteration in the relationship between total drug concentration and pharmacodynamic effect. For example, protein binding of phenytoin (90% protein-bound, primarily to albumin) is significantly reduced and this change alters the relationship between total phenytoin concentration and desired and toxic effects.8,38 The resulting increase in unbound fraction, from values of 10% in those with normal renal function to approximately 20% or more in those with stage 5 CKD, results in increased hepatic clearance and decreased total concentrations. Thus, in patients with CKD, the therapeutic range based on total phenytoin concentration is shifted downward from normal values of 10 to 20 mg/L (10 to 20 mcg/mL; 40 to 79 mmol/L) to values as low as 4 to 8 mg/L (4 to 8 mcg/mL; 16 to 32 mmol/L). Since the unbound concentration therapeutic range is the same for all patients, 1 to 2 mg/L (1 to 2 mcg/mL; 4 to 8 mmol/L), such a measurement provides the best target for individualizing phenytoin therapy in patients with CKD.
The unbound fraction of many acidic drugs increases in CKD patients as the result of a decrease in protein binding and this is associated with an increase in the apparent VD.8 A new equilibrium is ultimately established as a result of increased drug elimination/distribution, such that the unbound concentrations remain comparable to those observed in patients with normal renal function despite the fact that total concentrations are reduced. Thus, the net effect is an alteration in the relationship between total drug concentration and pharmacodynamic effect. For example, protein binding of phenytoin (90% protein-bound, primarily to albumin) is significantly reduced and this change alters the relationship between total phenytoin concentration and desired and toxic effects.8,38 The resulting increase in unbound fraction, from values of 10% in those with normal renal function to approximately 20% or more in those with stage 5 CKD, results in increased hepatic clearance and decreased total concentrations. Thus, in patients with CKD, the therapeutic range based on total phenytoin concentration is shifted downward from normal values of 10 to 20 mg/L (10 to 20 mcg/mL; 40 to 79 mmol/L) to values as low as 4 to 8 mg/L (4 to 8 mcg/mL; 16 to 32 mmol/L). Since the unbound concentration therapeutic range is the same for all patients, 1 to 2 mg/L (1 to 2 mcg/mL; 4 to 8 mmol/L), such a measurement provides the best target for individualizing phenytoin therapy in patients with CKD.
The decrease in plasma protein binding of acidic drugs (e.g., phenytoin) has been attributed to qualitative changes in the binding sites, accumulation of endogenous inhibitors of binding, and/or decreased concentrations of albumin. The first two of these mechanisms appear to account for most of the observed changes in binding. In addition, the high concentrations of metabolites of some compounds that accumulate in patients with end-stage renal disease (ESRD) may compete for protein binding sites with the parent compound.39
The principal binding protein for several basic drugs is α1-acid glycoprotein, an acute-phase reactant protein whose plasma concentrations are increased in CKD patients.4,8 As a result of this increase, the unbound fraction of some basic drugs (e.g., bepridil, disopyramide) may be significantly decreased and the VD increased in CKD patients, especially renal transplant and hemodialysis patients.4,8
Effect of Altered Tissue Binding
Altered tissue binding may also result in a reduction of the apparent VD of a drug. For example, the VD of digoxin has been reported to be reduced by 30% to 50% from the normal values in patients with stage 5 CKD, as well as in hemodialysis patients.40 This reduction in VD may be a result of decreased tissue binding, the presence of acidosis, or digoxin-like immunoreactive substances.9,41 In this case, the absolute amount of digoxin bound to the receptor is reduced and the resultant serum digoxin concentration is higher than anticipated. Thus, in patients with renal insufficiency, particularly in those with stage 5 CKD, a “normal” total drug concentration may be associated with either an adverse reaction secondary to elevated unbound drug concentrations, or a subtherapeutic response because of an altered plasma-to-tissue drug concentration ratio. The monitoring of unbound drug concentrations in CKD patients is thus warranted for those drugs that have a narrow therapeutic range, are highly protein bound (free fraction of <20%), and for which marked variability in the free fraction has been reported (e.g., phenytoin and disopyramide).
Effect of VD Calculation Method
Finally, the method used to calculate the volume of distribution may be influenced by renal insufficiency. The three most commonly used volume of distribution terms are: volume of the central compartment (Vc), volume of the terminal phase (Vβ and Varea), and volume of distribution at steady state (Vss). The Vc for many drugs approximates extracellular fluid volume and thus may be increased or decreased by acute changes. Oliguric acute renal failure is often accompanied by fluid overload and a resultant increased Vc for many drugs. The Varea or Vβ represents the proportionality constant between plasma concentrations in the terminal elimination phase and the amount of drug remaining in the body. Vβ is affected by both distribution characteristics, as well as by the terminal elimination rate constant. Vβ and Vss will often be similar in magnitude, with Vβ being slightly larger. Because Vss has the advantage of being independent of drug elimination, it is the most appropriate volume term to use when one desires to compare drug distribution volumes between patients with renal insufficiency and those with normal renal function.42
Drug Metabolism
Accumulation of Metabolites
There are only a few drugs that are eliminated completely unchanged via the kidneys and thus most drugs are metabolized to some extent. Patients with severe renal insufficiency who are receiving chronic drug therapy may experience accumulation of metabolite(s) as well as the parent compound. Metabolites of several drugs have been reported to have significant pharmacologic and/or toxicologic activity.43 However, the pharmacokinetics and pharmacodynamics of metabolites are not often fully elucidated. In a sense, the patient with severe CKD is being exposed to a new pharmacologic entity since the sum of the serum concentrations of the metabolite and the parent compound are markedly different than those reported in patients with normal renal function.
The metabolite may have pharmacologic activity similar to that of the parent drug and thus contribute significantly to clinical response; that is true, for example, of oxypurinol. Alternatively, the metabolite may have qualitatively dissimilar pharmacologic action; for example, normeperidine has CNS stimulatory activity that reportedly produces seizures, whereas meperidine has CNS depressant actions.44,45 Because of the multiplicity of potential interactions of compounds that are primarily metabolized, the practical consequences of metabolite accumulation are difficult to predict and are most often identified in those patients at risk by trial and error.
Alterations of CYP 450 Enzyme Activity
![]() A decrease in the renal clearance of drugs in patients with CKD is well appreciated. However, there is now good preclinical and emerging clinical evidence which suggests that CKD may lead to alterations in nonrenal clearance of many medications as the result of alterations in the activities of uptake and efflux transporters as well as CYP enzymes in the liver and other organs8–12,46 (Table 33-2). The effect(s) of renal insufficiency on nonrenal drug clearance appear to depend on whether the reduction in renal function is acute or chronic in nature. For example, higher residual nonrenal clearance for vancomycin, meropenem, and imipenem has been documented in patients with AKI compared to patients with CKD, who have comparable CLcr.47 In humans with renal insufficiency, the activities of CYPs appear to be relatively unaffected. It was reported that CYP3A4 activity was reduced,7,9–12 but recent data indicate that organic anion-transporting polypeptide (OATP) uptake activity is reduced and thus the perceived changes in CYP3A4 activity were likely due to altered transporter activity, not an alteration in CYP activity. The reduction of nonrenal clearance of several drugs that exhibit overlapping CYP and transporter substrate specificity in patients with stage 4 or stage 5 CKD supports this premise (Table 33-3). These studies must be interpreted with caution, however, because concurrent drug intake, age, smoking status, and alcohol intake were often not taken into consideration. Furthermore, pharmacogenetic variations in drug-metabolizing enzymes that may have been present in the individual before the onset of AKI or CKD must be considered, if known.7,9–12 This differential effect on individual enzymes may help to explain some of the conflicting reports of whether drug metabolism is altered in the presence of CKD. Cytochrome P450 enzyme 3A activity as measured by the erythromycin breath test (EBT) is 28% lower in ESRD patients as compared with healthy controls.48 Although baseline CYP3A activity was lower in these patients, the increase in CYP3A activity observed following enzyme induction with rifampin was similar.48 Nolin and colleagues have recently shown that EBT results are reduced more in those ESRD patients with higher blood urea nitrogen concentrations and that hemodialysis is associated with an acute improvement in the patient’s metabolic activity.10 These data indicate that CKD has a detrimental effect on this important pathway of hepatic drug metabolism in humans.
A decrease in the renal clearance of drugs in patients with CKD is well appreciated. However, there is now good preclinical and emerging clinical evidence which suggests that CKD may lead to alterations in nonrenal clearance of many medications as the result of alterations in the activities of uptake and efflux transporters as well as CYP enzymes in the liver and other organs8–12,46 (Table 33-2). The effect(s) of renal insufficiency on nonrenal drug clearance appear to depend on whether the reduction in renal function is acute or chronic in nature. For example, higher residual nonrenal clearance for vancomycin, meropenem, and imipenem has been documented in patients with AKI compared to patients with CKD, who have comparable CLcr.47 In humans with renal insufficiency, the activities of CYPs appear to be relatively unaffected. It was reported that CYP3A4 activity was reduced,7,9–12 but recent data indicate that organic anion-transporting polypeptide (OATP) uptake activity is reduced and thus the perceived changes in CYP3A4 activity were likely due to altered transporter activity, not an alteration in CYP activity. The reduction of nonrenal clearance of several drugs that exhibit overlapping CYP and transporter substrate specificity in patients with stage 4 or stage 5 CKD supports this premise (Table 33-3). These studies must be interpreted with caution, however, because concurrent drug intake, age, smoking status, and alcohol intake were often not taken into consideration. Furthermore, pharmacogenetic variations in drug-metabolizing enzymes that may have been present in the individual before the onset of AKI or CKD must be considered, if known.7,9–12 This differential effect on individual enzymes may help to explain some of the conflicting reports of whether drug metabolism is altered in the presence of CKD. Cytochrome P450 enzyme 3A activity as measured by the erythromycin breath test (EBT) is 28% lower in ESRD patients as compared with healthy controls.48 Although baseline CYP3A activity was lower in these patients, the increase in CYP3A activity observed following enzyme induction with rifampin was similar.48 Nolin and colleagues have recently shown that EBT results are reduced more in those ESRD patients with higher blood urea nitrogen concentrations and that hemodialysis is associated with an acute improvement in the patient’s metabolic activity.10 These data indicate that CKD has a detrimental effect on this important pathway of hepatic drug metabolism in humans.
TABLE 33-2 Impact of ESRD on Nonrenal Clearance of Selected Drugs
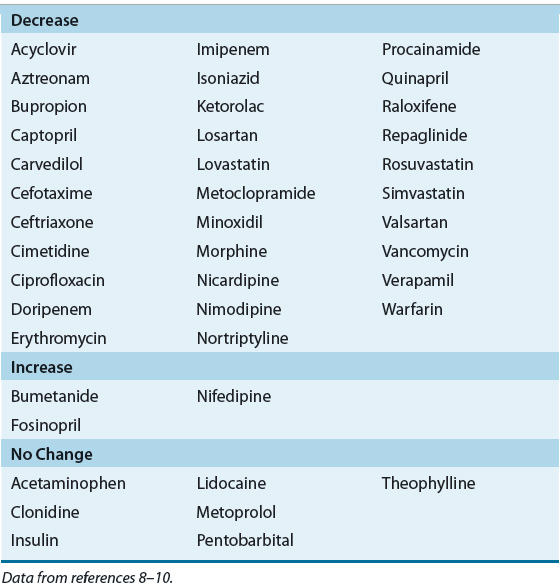
TABLE 33-3 Major Pathways of Nonrenal Drug Clearance
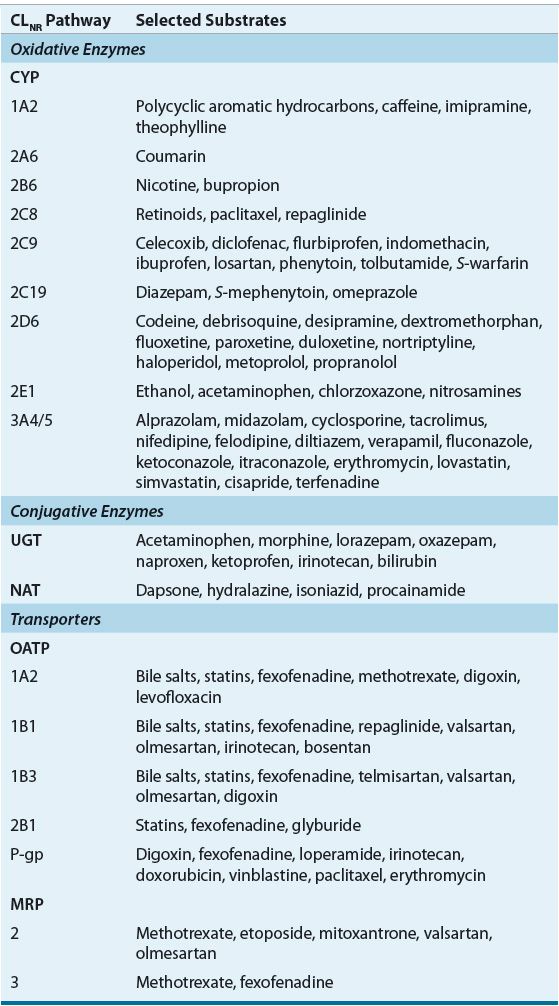
Prediction of the effect of renal insufficiency on the metabolism of a particular drug is however difficult and there is currently no quantitative strategy to predict changes for one drug based on data from another even if they are in the same pharmacologic class. However, some qualitative insight can be gained if one knows what enzyme is involved in the metabolism of the drug of interest and how the enzyme(s) or transporter(s) is affected by the presence of CKD.
EFFECT OF CHRONIC KIDNEY DISEASE ON RENAL TRANSPORTERS
Renal clearance (CLR) of a drug is the composite of GFR, tubular secretion, and reabsorption (CLR = [GFR × fu] + [CLsecretion – CLreabsorption]), where fu is the fraction of the drug unbound to plasma proteins. Drug elimination by filtration occurs by diffusion; while tubular secretion and reabsorption are bidirectional processes that involve carrier-mediated renal transport systems.49 Renal transport systems have been broadly classified on the basis of substrate selectivity into the anionic and cationic renal transport systems, which are responsible for the transport of a number of organic acidic and basic drugs, respectively (see eChap. 18 and Table 33-3).8 Several drugs are actively secreted by one or more of these transporter families, which include organic cationic (e.g., famotidine, trimethoprim, and dopamine), organic anionic (e.g., ampicillin, cefazolin, and furosemide), nucleoside (e.g., zidovudine), and P-glycoprotein transporters (e.g., digoxin, vinca alkaloids, and steroids).50,51 Alterations in filtration, secretion, or reabsorption, secondary to CKD may have a dramatic effect on drug disposition: for drugs that are primarily filtered, a reduction in GFR will result in a proportional decrease in renal drug clearance.
ESTIMATION OF RENAL FUNCTION
In the absence of data delineating the contribution of tubular function to renal clearance, the clinical measurement or estimation of CLcr or GFR remains the guiding factor for drug dosage regimen design.4,6,12 The importance of an alteration in renal function on drug elimination depends on two factors: the fraction of drug normally eliminated by the kidney unchanged and the degree of renal insufficiency. Quantitation of the patient’s renal function can be accomplished by measurement of CLcr or GFR or estimation of CLcr or GFR based on the stable serum creatinine concentration (see eChap. 18). Because of the time delay involved and problems in obtaining complete urine collections, measured CLcr or GFR values are infrequently used for initial drug dosage regimen design. Therefore the calculation of initial drug dosage regimens relies on the estimation of CLcr or GFR in adults and children from such routinely available clinical data as age, gender, height, weight, and serum creatinine or serum cystatin C. The best method to use for adults remains controversial with many supporters of the C–G equation and others who support the use of the MDRD or chronic kidney disease epidemiology collaboration equation (CKD-EPI) approach52–54 (see eChap. 18).
Recently, there is considerable debate regarding the use of C–G or MDRD equation to guide drug dosing adjustment in patients with CKD. Recommendations as to when to adjust drug dosage regimens for currently approved medications are largely (greater than 95%) based on relationships between drug clearance and CLcr calculated by the C–G equation.55 Several studies have shown that when compared to drug dosing based on C–G, the use of MDRD equation resulted in conflicting drug dosing recommendations in 40% of patients.56–58 A recent systematic comparison between C–G and MDRD equations showed that the classification of renal function with each equation was generally similar.59 However, using the MDRD equation resulted in conflicting drug dosing recommendations (higher or lower dosing categories) in 12% of patients and resulted in higher dosing recommendations in patients with a combination of advanced age (age > 80 years), low weight (<55 kg), and modest elevations of serum creatinine (>0.7 and ≤1.5 mg/dL [>62 and ≤133 μmol/L]).59
The introduction of the 2009 CKD-EPI creatinine equation complicates things further.60 Reports showed that the CKD-EPI creatinine equation was more accurate in those with higher mean measured GFRs (estimated to be >60 mL/min per 1.73 m2); however, the MDRD equation was more accurate in those with GFRs less than 60 mL/min per 1.73 m2.52 In 2012, there were two variations of the CKD-EPI equation (CKD-EPI cystatin C equation and CKD-EPI creatinine–cystatin C equation) reported, which utilize cystatin C with or without serum creatinine to estimate GFR. One study showed that the CKD-EPI creatinine-cystatin C equation performed better and resulted in more accurate estimates of GFR compared to equations that used creatinine or cystatin C alone (CKD-EPI cystatin C or CKD-EPI creatinine equations).54 Although, recent data show improved accuracy of estimating GFR, especially with the CKD-EPI creatinine-cystatin C equation, there is extremely little data regarding the use of these equations to guide drug dosing in CKD patients. As such, drug dosing recommendations should continue to be based on CLcr estimated by C–G for the present.55
DRUG DOSAGE REGIMEN DESIGN FOR PATIENTS WITH CHRONIC KIDNEY DISEASE
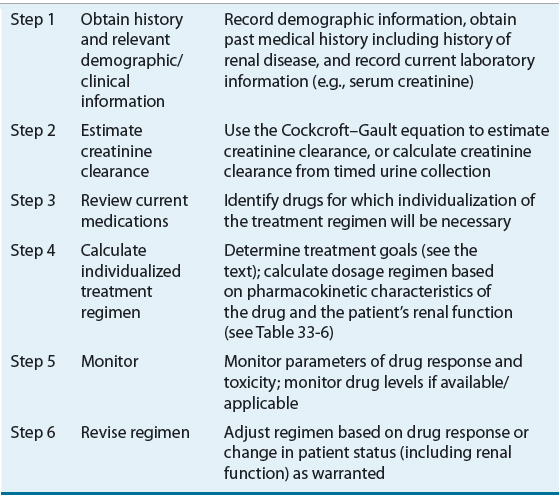
![]() The initial or “loading” dose for should be the same for patients with impaired renal function as those with normal renal function unless the volume of distribution is known to be altered in the presence of renal insufficiency or a concomitant disease (Table 33-1). Maintenance dosage regimen guidelines for CKD patients in FDA or European Medicines Agency (EMA) approved product labeling should be the foundation for ongoing therapy. If such information is not available or if there is marked variance between these two agencies’ recommendations the approach depicted in Table 33-4 for designing a dosage regimen for a patient with CKD can be utilized. In either case the design of the optimal dosage regimen is dependent on the availability of an accurate characterization of the relationship between the pharmacokinetic parameters of the drug and renal function and an accurate assessment of the patient’s renal function.
The initial or “loading” dose for should be the same for patients with impaired renal function as those with normal renal function unless the volume of distribution is known to be altered in the presence of renal insufficiency or a concomitant disease (Table 33-1). Maintenance dosage regimen guidelines for CKD patients in FDA or European Medicines Agency (EMA) approved product labeling should be the foundation for ongoing therapy. If such information is not available or if there is marked variance between these two agencies’ recommendations the approach depicted in Table 33-4 for designing a dosage regimen for a patient with CKD can be utilized. In either case the design of the optimal dosage regimen is dependent on the availability of an accurate characterization of the relationship between the pharmacokinetic parameters of the drug and renal function and an accurate assessment of the patient’s renal function.
TABLE 33-4 Stepwise Approach to Adjust Drug Dosage Regimens for Patients with Renal Insufficiency