A drug-nutrient interaction is defined as the result of a physical, chemical, physiologic, or pathophysiologic relationship between a drug and nutrient status, a nutrient, multiple nutrients, or food in general (1). The causes of most clinically significant drug-nutrient interactions are usually multifactorial. Failure to identify and properly manage drug-nutrient interactions can lead to very serious consequences (2). In the case of treating an infection, for instance, some drug-nutrient interactions can result in reduced absorption of certain oral antibiotics and lead to suboptimal antibiotic concentrations at the site of infection (2, 3). This predisposes the patient to treatment failure and antibiotic resistance in the future. A drug-nutrient interaction also may become a burden on health care costs if its associated complications, such as treatment failure or adverse events, result in increased length of hospital stay and overall resource utilization. In some instances, the impacts of unrecognized and unmanaged drug-nutrient interactions may take years to surface. Metabolic bone disease secondary to vitamin D deficiency in transplant recipients or patients with epilepsy usually takes months to years to progress (4, 5, 6, 7, 8). Without proper monitoring of mineral and vitamin status and early intervention, accelerated bone loss or bone fracture may occur.
PREDICTORS
Research has shown that the main predictors for having a drug-nutrient interaction in a given patient are increased age, presence of multiple chronic illnesses, and the concurrent use of multiple medications and supplements (9). Critically ill patients—especially those who receive continuous enteral feeding—may also be at risk for developing drug-nutrient interactions (10, 11). The presence of polypharmacy (i.e., using multiple drugs to manage different disease states) further increases patients’ risk for problematic drug-nutrient interactions (12). Elderly patients, patients who have underlying malnutrition, or obese patients are more likely to experience more severe adverse events from a drug-nutrient interaction because of the changes in body composition and physiologic reserves (13, 14, 15, 16, 17, 18). Patients with multiple chronic diseases are also more prone to having adverse events. Genetics can be an important factor in determining the appropriate dose and clinical response to a particular drug or nutrient. Polymorphism of the methylenetetrahydrofolate reductase gene (MTHFR) may affect the amount of pyridoxine, cobalamin, folic acid, and riboflavin requirement, which may play a role in determining the threshold intake in preventing certain drug-nutrient interactions (19, 20, 21). Although the clinical significance has not been well documented, it is possible that some genetic polymorphisms also may have a protective effect against experiencing clinically significant drug-nutrient interactions.
CLASSIFICATION
A drug-nutrient interaction may result in an alteration of the kinetic or dynamic profile of a drug or a nutrient (22). The magnitude of change determines whether the interaction is clinically significant and requires intervention. Pharmacokinetics refers to the quantitative description of drug disposition, which includes absorption, distribution, metabolism, and excretion of the compound. Pharmacokinetic parameters such as half-life, bioavailability, time to reach maximal detectable concentration (tmax), and area-under-the-concentration-time curve (AUC) often are used to provide quantitative comparisons. Half-life refers to the time it takes for the drug concentration (usually in the plasma) to reduce by one-half. It is used commonly to reflect the rate of removal or clearance of the drug from the body. Bioavailability refers to the fraction of the drug administered that becomes available in the body. By definition, intravenous administration provides 100% bioavailability. Oral administration, in many cases, produces lower bioavailability because of incomplete absorption or loss of the active component from presystemic effect. Oral bioavailability is usually the most significant parameter affected by drug-nutrient interactions, although the rate of absorption, metabolic clearance, and tissue distribution of compounds may also be altered. The parameter tmax is used to determine the time to achieve peak plasma concentration of a particular compound. If the drug or nutrient is administered orally, tmax reflects the rate of oral absorption. Age, underlying medical conditions, types of diet consumed, surgical interventions to the gastrointestinal tract, and concurrent medications can all affect tmax. The parameter AUC is used to reflect the overall exposure of a drug by the patient. It is affected by oral bioavailability, clearance and, in some cases, the rate of absorption. The disposition and kinetics of a nutrient (i.e., nutrikinetics) can also be described by these mathematical parameters. For instance, one can estimate the effect of a drug on the disposition of vitamin D by comparing the bioavailability, distribution, and elimination rate of calcidiol and calcitriol before and after the introduction of the drug.
Pharmacodynamics refers to the clinical or physiologic effects of the drug. For example, coadministration of folic acid to a patient taking the antiepileptic agent phenytoin may lead to a reduction in serum phenytoin concentration (pharmacokinetic effect). If the reduction in phenytoin concentration is clinically significant, the patient may experience increased frequency or duration of seizure activities (pharmacodynamic effect).
The four types of drug-nutrient interactions are categorized based on the nature and the mechanisms of the interactions (22). Each type is described briefly in the following paragraphs, and the terms “object agent” and “precipitant agent” are used. An object agent refers to the drug or the nutritional element that is affected by the interaction. A precipitant agent refers to the drug or the nutritional element that causes the interaction.
Type I: Ex vivo bioinactivations, which refers to the interaction between the drug and the nutritional element or formulation through biochemical or physical reactions. Some of the examples of this type of interaction involve hydrolysis, oxidation, neutralization, precipitation, and complexation. These reactions usually take place when the interacting agents are in direct physical contact and occur usually before the nutrients or drugs enter the body. In other words, these interactions usually occur in the delivery device.
Type II: Interactions affecting absorption, which affect drugs and nutrients delivered only by mouth or via enteral delivery devices. These interactions cause either an increase or decrease of oral bioavailability of the object agent. The precipitant agents may modify the function of an enzyme (type A interaction) or a transport mechanism (type B interaction) that is responsible for the biotransformation or transport of the object agent before reaching systemic circulation. In some cases, complexation, binding, or other deactivating processes occur in the gastrointestinal (GI) tract (type C interaction) and impair the object agent from being absorbed.
Type III: Interactions affecting systemic/physiologic disposition, which occur after the drug or the nutritional element has been absorbed from the gastrointestinal tract and entered into the systemic circulation. The mechanisms involve changing the cellular or tissue distribution, systemic metabolism or transport, or penetration to specific organ/tissues of the object compound. In some cases, the interaction between the precipitant agent and the object agent may involve changing the function of other cofactors (e.g., clotting factors) or hormones.
Type IV: Interactions affecting the elimination or clearance of drugs or nutrients, which may involve the modulation, antagonism, or impairment of renal or enterohepatic elimination.
FACTORS AFFECTING DRUG-NUTRIENT INTERACTIONS
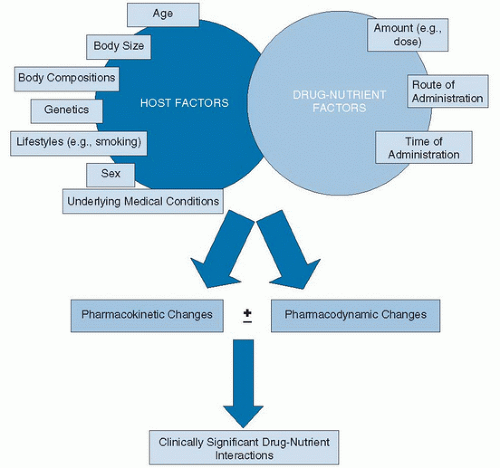
The two factors that play the most significant role in the occurrence of drug-nutrient interactions are the host and the drug (or nutrient) itself (Fig. 103.1). The host factor refers to the individual’s response to a drug or nutrient. Age, sex, body size, body composition, lifestyle, underlying diseases and medical conditions, and genetics can affect the response (Table 103.1). The drug or nutrient factor is always affected by the amount, time, and route of administration. For instance, type II drug-nutrient interaction occurs exclusively when both the nutrient and the drug are administered orally or via enteral route and can essentially be eliminated by administration via the intravenous route. Many type IIC interactions (complexation in the GI tract) can be avoided by simply spacing out the administration time. Type I interactions are far more common with drugs and nutrients administered intravenously. The potential for drug-nutrient interactions increases exponentially with the addition of every drug or dietary supplement (23).
Fig. 103.1. Factors contributing to drug-nutrient interactions. Not all pharmacokinetic or pharmacodynamic changes may lead to clinically significant interactions.
MECHANISM
Ex Vivo Bioinactivations
Before reaching the GI tract or the venous blood (as in the case of intravenous administration), drugs and nutrients are sometimes mixed in the delivery system, where physicobiochemical reactions may occur ex vivo between drugs and nutrients in a delivery vehicle or tubing, in which the active ingredients can be deactivated before being absorbed. The resultant biophysical change as the result of the interaction is described as physical incompatibility. This commonly happens with parenteral products, because physical incompatibility is often associated with the formation of precipitates in a solution. Some of the physical incompatibility can even be visually detected. Precipitation from calcium and phosphate salts is a classic example of physical incompatibility resulting in the formation of the insoluble particles. Nevertheless, visual inspection is not the most accurate approach to rule out the presence of physical incompatibility because some of the precipitants formed may be too small to be visible by naked eyes (24). Intravenous lipid products, conversely, are formulated as opaque emulsions (oil mixed in water). Therefore, physical incompatibility cannot be determined by visual inspection. As a general rule, practitioners should not rely on visual inspection to determine the presence of type I interactions (Table 103.2).
TABLE 103.1 PATIENT POPULATIONS WITH SIGNIFICANTLY HIGHER RISK OF EXPERIENCING ADVERSE EVENTS FROM DRUG-NUTRIENT INTERACTIONS
Patients with acquired immunodeficiency syndrome
Patients with cancer
Elderly patients
Malnourished patients
Patients with gastrointestinal tract dysfunctions or surgery (e.g., bariatric surgery, Crohn disease)
Patients receiving enteral nutrition
Pregnant women
Transplant recipients
TABLE 103.2 SUGGESTED APPROACHES TO MINIMIZE TYPE I DRUG-NUTRIENT INTERACTION
Do not mix drugs directly with feeding formulas (enteral or parenteral).
Hold enteral feeding before administering drugs through enteral feeding tube; holding time varies from a few minutes to 2 hours. Feeding rate should be adjusted accordingly to prevent underfeeding. Signs and symptoms of feeding intolerance should be monitored.
Flush the feeding tube with water before and after drug administration.
Use preformulated oral solutions, elixirs, or suspensions instead of crushing tablets when administering drugs through enteral feeding tubes, to increase dose accuracy and minimize clogging.
For parenteral agents, always consult with pharmacists or references to determine their compatibility.
Do not rely on visual inspection to determine compatibility among intravenously administered products; use published references.
Consider monitoring drug levels for drugs with narrow therapeutic indices.
Effect of Meal Intake on Drug and Nutrient Absorption
Concurrent meal intake and drug administration can influence oral absorption of drugs and nutrients, and is an example of type II interactions. The overall impact may include changing the rate of absorption, changing the magnitude of absorption, or both. The mechanisms affecting drug or nutrient absorption with concurrent meal intake usually involve one or more of the following factors: (a) altering gastric acid and gastrin secretion, (b) altering gastrointestinal transit time, (c) changing dissolution of drugs in solid dosage forms, (d) binding or complexation of micro- or macronutrient with food contents, and (e) changing bile flow (25, 26, 27).
Meal intake generally stimulates gastric and intestinal secretions (28, 29), which theoretically favor drug dissolution from their solid dosage form and facilitate absorption. Meals with higher fat content stimulate the release of bile salts, which also facilitate the intestinal uptake of drugs that are more lipophilic (30). Furthermore, high-fat meals promote the release of cholecystokinin, which slows GI motility and increases the contact time between the drug molecules and the intestinal epithelial tissues (31). All these factors combined tend to favor a more complete absorption of certain drugs and nutrients. For example, the oral bioavailability of albendazole and griseofulvin is dramatically increased when taken with a fatty meal (32, 33). However, the potential physicochemical interactions, the potential binding affinity between the individual drug and food contents, the dose of the drug administered, and the composition of the meals make drug absorption in the presence of food rather erratic and unpredictable. Therefore, concurrent food intake actually contributes to the variation in the magnitude of the drug and nutrient interaction observed (26). For instance, the data on how food affects the oral bioavailability of verapamil, a calcium channel blocker used in the treatment of hypertension and cardiac arrhythmias, are very inconsistent. A small reduction of verapamil oral bioavailability with concurrent food intake was seen in some studies, whereas a similar number of studies failed to show any clinically relevant change in verapamil bioavailability. Overall, the AUCs of verapamil observed were similar whether it was taken with or without food. The only consistent finding among the studies was that food slowed down the rate of verapamil absorption (34, 35, 36, 37). These findings suggest that verapamil can be taken without regard to food, as long as it is on a consistent basis.
It is important to differentiate between delayed absorption versus decreased absorption. A delayed absorption of a drug by food does not necessarily lead to a reduced absorption. In many cases, food may impair the rate of drug absorption, as measured by the time required to reach the highest serum drug concentration (or tmax) without affecting the total amount absorbed, as measured by AUC. Delayed absorption of drugs caused by food may be explained by the highly variable physiologic response to feeding among individuals, as well as the inconsistency in food contents consumed between meals. Drugs such as famciclovir, methotrexate, verapamil, levetiracetam, and levodopa have delayed absorption in the presence of food, as measured by increased tmax. However, the total amount absorbed is comparable with or without food, as quantified by the AUCs (38, 39, 40, 41, 42, 43, 44). In these cases, food can delay the onset of the drug action; however, the efficacy should not be affected. Despite a detectable difference in the pharmacokinetic parameter, these drug-nutrient interactions may not be considered clinically significant. On the contrary, if food intake causes a significant change in the AUC of a drug, it implies that the overall amount absorbed is reduced. This kind of interaction is more clinically significant, and pharmacodynamic changes may be detected. In this case, patients should receive specific instructions on taking the drug with regard to meal intake. Medication adherence should be emphasized and monitored to warrant stable and consistent drug responses and prevent undesired effects or therapeutic misadventures.
The presence of food causes distention of the stomach and stimulates the autonomic nervous system to alter GI tone (45, 46). Together with the endocrinologic changes induced by the food contents (e.g., releases of insulin, cholecystokinin, gastrin), splanchnic blood flow is also increased (47, 48, 49). Increased blood delivery to the liver through the hepatic portal vein can augment presystemic metabolism. However, since the metabolism of most drugs depends primarily on the host’s intrinsic clearance (i.e., the amount and the availability of the drug metabolizing enzymes) but not hepatic blood flow, food-induced augmentation of splanchnic and hepatic blood flow rarely causes clinically significant drug-nutrient interaction. One exception is ethanol, a compound with high extraction ratio whose metabolism appears to be highly affected by hepatic blood flow. Concurrent food intake with 530 kcal increased the presystemic metabolism of ethanol by up to 49%, with men showing more dramatic increase than women (50). Hepatic blood flow can be estimated by the use of intravenous indocyanine green (51). Tables 103.3 and 103.4 summarize the commonly prescribed medications that should be taken with food or on an empty stomach, respectively.
TABLE 103.3 DRUGS THAT ARE RECOMMENDED TO TAKE WITH FOOD TO MAXIMIZE ABSORPTION
Albendazole
Ketoconazole
Amiodarone
Lithium
Atazanavir
Lopinavir
Atovaquone
Lovastatin
Cefuroxime
Mefloquine
Erythromycin ethylsuccinate
Nelfinavir
Ganciclovir
Rifapentine
Griseofulvin
Ritonavir
Hydralazine
Saquinavir
Itraconazole (capsule only)
In summary, whether a drug should be taken with food or on an empty stomach depends mostly on the characteristics of the drug. When in doubt, data and results from the primarily literature should be used to guide clinical decisions. Drugs with decreased oral bioavailability in the presence of food, as suggested by a reduction of AUC, should be taken on an empty stomach. Drugs with delayed, but not decreased, absorption in the presence of food, as described by a change in tmax but no change to AUC, may be taken without regard to meal intake.
Modulation of Gastrointestinal Motility
Gastric emptying and intestinal transit time may alter the rate and magnitude of drug or nutrient absorption from the GI tract. GI motility is regulated by four major components—neurotransmitters present in the enteric nervous system such as acetylcholine, serotonin, and dopamine; gut peptides such as ghrelin, type 1 glucagon-like peptide (GLP1); electrical activity such as pacing and regulation by the interstitial cell of Cajal; and serum blood glucose concentration (52, 53). Motility of the upper GI tract is increased with hunger. Continued food consumption leads to the release of inhibitory neuroendocrines and peptides, such as GLP1, and cholecystokinin that inhibit gastric emptying and eventually cause satiety (54, 55, 56). Decreased GI motility increases the contact time for the drug or nutrient with the GI epithelial tissue, thus potentially allows more complete absorption of the drug molecules or nutrients into the portal vein.
TABLE 103.4 DRUGS THAT SHOULD BE TAKEN ON AN EMPTY STOMACH TO MAXIMIZE ABSORPTION
c Chelation and/or binding with food content or divalent and trivalent cations (e.g., calcium magnesium).
d Food may deactivate the drug through precipitation.
The enteric nervous system affects both motility and secretions in the GI tract. Increased GI secretion provides a complementary effect on absorption and the digestive process (57, 58). Vagal stimulation increases upper GI contraction and decreases pyloric sphincter contractions. As a result, gastric emptying is promoted and the time required for the drug or nutrient to reach the small intestine is shortened. Decreased gastric emptying time may reduce drug absorption because of less complete dissolution (for solid dosage forms) and shortened physical contact time between the drug molecules and the epithelial tissues. Conversely, drug and nutrient absorption may be increased by the coadministration of opioid derivatives, which slow GI motility, or drugs with anticholinergic effect (e.g., sedating antihistamines, phenothiazines, tricyclic antidepressants) that block vagus nerves. However, anticholinergic effects may also decrease secretion in the intestine and affect nutrient absorption. Overall, significantly decreased GI tone may precipitate toxicities because of more complete absorption, whereas elevated GI motility may increase risk of therapeutic failure.
Only gold members can continue reading. Log In or Register to continue