Dissolution testing of solid dosage forms
Ana Cristina Freire and Abdul W. Basit
Chapter contents
The relevance of drug dissolution and dissolution testing
General requirements for in-vitro dissolution testing
The design of suitable dissolution tests; QC versus predictive dissolution testing
Dissolution testing for quality control
Compendial dissolution apparatuses
Volume and composition of the dissolution medium
Predictive dissolution testing
Bio-relevant dissolution media
Key points
• Dissolution testing is a key quality control test.
• Dissolution tests are normally performed under sink conditions.
The relevance of drug dissolution and dissolution testing
The biological events that occur in the body following the administration of a drug are varied and complex. Before being absorbed into the bloodstream, an orally administered drug is exposed to the dynamic conditions of the gastrointestinal lumen and much of what happens here will have a bearing on its therapeutic activity.
Only drug in its dissolved state can permeate the intestinal mucosa. If the solubility of the drug in the gastrointestinal aqueous fluids is limited, dissolution will be the rate-limiting step to absorption, and will dictate the rate and extent to which the drug becomes available in the bloodstream. Because of the link between oral bioavailability and dissolution, most oral drug products such as suspensions, granules, pellets, tablets and capsules are currently required to be tested for their dissolution characteristics. Disintegration measurements are also routinely performed on immediate-release products, although, for the most part, the results from such tests correlate poorly with bioavailability.
The concept of dissolution from a physical chemistry standpoint is well established; research into the mechanisms of dissolution of solids in liquids started over a century ago with the pioneering work of Noyes and Whitney (discussed fully in Chapter 2). However, it took several decades before the link between dissolution and oral bioavailability in the context of a pharmaceutical product was fully appreciated. This is considered to have occurred in 1951, when Edwards studied the dissolution of aspirin tablets in different media. His conclusions from that study were that “the dissolution of an aspirin tablet in the stomach and intestine is the rate process controlling the absorption of aspirin into the bloodstream”.
The clinical relevance of Edward’s observations was only realized in the mid-1960s, when several reports linking dissolution data with clinical efficacy, inferior efficacy and even toxicity of some commercial oral solid products were made available. For example, clinical inadequacies with formulations of the oral anti-diabetic and poorly water soluble drug, tolbutamide, were documented. Ineffective tablets were shown to disintegrate and dissolve at a slower rate than clinically effective ones (Fig. 35.1).
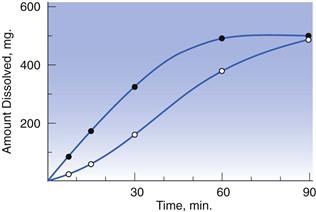
Fig. 35.1 Dissolution of tolbutamide from clinically effective (–•–) and ineffective (– –) tablets, as a function of time. (Courtesy of Levy, 1964, with permission.)
–) tablets, as a function of time. (Courtesy of Levy, 1964, with permission.)
Reports of toxicity with a formulation of phenytoin available in Australia and New Zealand followed in 1968. The substitution of the more hydrophilic excipient lactose for calcium sulfate in an immediate-release hard gelatin capsule enhanced the dissolution rate and bioavailability of phenytoin, leading to numerous cases of anticonvulsant intoxication. This is further discussed in Chapter 33 (see Fig. 33.6 and associated text).
However, undoubtedly the reference case for the impact of drug dissolution on oral bioavailability was that of digoxin tablets. In the early 1970s, several marketed formulations of digoxin were shown to yield up to seven-fold differences in blood levels of the drug. In search for an explanation, digoxin tablet formulations were studied for their dissolution properties, exposing huge differences in this parameter. Interestingly, a good correlation was noted between dissolution rate and digoxin blood levels. In other words, absorption of digoxin was higher for those formulations which dissolved quicker (Fig. 35.2). In contrast, the disintegration time of the different tablets was very similar and bore no relation to digoxin blood levels, raising questions as to the value of the disintegration test in detecting differences in the oral bioavailability of solid drug products.
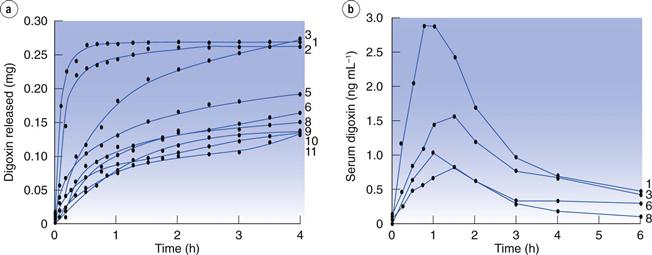
Fig. 35.2 Dissolution rates of different brands of digoxin tablets available in the UK market at the time of the study (a) and corresponding blood (serum) levels of digoxin (b). Formulation 1 (‘new Lanoxin’) and 8 (‘old Lanoxin’) are from the same brand, prepared using different manufacturing methods. (Courtesy of Fraser et al, 1973.)
In light of these cases, it became clear that the formulation and manufacturing process was linked to the therapeutic efficacy of the drug. Dissolution, and not disintegration, became an accepted indicator of oral bioavailability of solid drug products. This prompted the Food and Drug Administration (FDA) in the United States of America to make the use of dissolution testing official in 1970, and to introduce dissolution requirements in the pharmacopoeial monographs of tablets and capsules. By 1995, four different dissolution apparatuses had been included in the United States Pharmacopeia (USP).
General requirements for in-vitro dissolution testing
Today, in-vitro dissolution studies are carried out for several reasons:
• most often to ensure that preparations comply with product specifications
• to provide an indication of the performance of the preparation in vivo.
Changes to the release profile and/or dissolution rate of the drug can be brought about by the characteristics of the dosage form and by its method of manufacturing. However, dissolution becomes more complicated if the properties of the physiology of the gastrointestinal tract are taken into consideration (Chapter 19). These must be reflected in any efficient in-vitro test.
pH of the gastrointestinal luminal fluids
As it travels through the gastrointestinal tract, a drug is exposed to increasing pH conditions. Such pH conditions play an important role in the solubility of ionizable drugs, with pKa values within the pH physiological range (1–7.5), and may affect their bioavailability (discussed fully in Chapter 20). This must be simulated, particularly in predictive dissolution testing (see below).
Composition of the gastrointestinal luminal fluids
While pH may be sufficiently simulated with buffered solutions, the composition of the gastrointestinal tract fluids is not. Gastrointestinal luminal fluids are enriched with amphiphilic bile components, such as bile salts and lecithin. These substances enhance the dissolution rate of drugs either via an increase in wettability or, at higher concentrations, through the formation of micelles. Although fasted fluids already have wetting and solubilizing effects, this is further increased after a meal, due to increased secretion of bile and to the presence of degradation products of lipids contained in the meal (i.e. fatty acids and monoglycerides). For this reason, the dissolution of poorly soluble drugs is generally higher under fed than in fasted conditions. This is well-exemplified by the lipophilic drug, danazol; as seen in Figure 35.3 the bioavailability of this compound is considerably higher in the fed state.
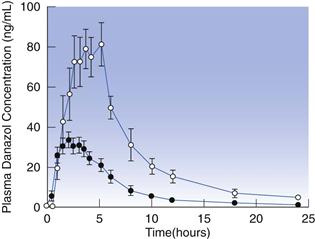
Fig. 35.3 Plasma concentration-time profiles following administration of 100 mg danazol in a hard gelatin capsule in the fed (– –) and fasted (–•–) states in humans. (Courtesy of Charman et al 1993.)
–) and fasted (–•–) states in humans. (Courtesy of Charman et al 1993.)
Two main uses of in-vitro dissolution testing are now discussed further.
As a quality control tool
Before a drug product is released onto the market place (and routinely once marketed), it must be subjected to rigorous control to ensure that the quality and performance (with regards to both safety and efficacy) of the final product are acceptable. Currently, almost all solid oral drug products require in-vitro dissolution testing as part of their quality control (QC) assessment. This involves analysing the drug release profile of different batches of the same drug product with a view to either confirm manufacturing and product consistency or to verify the stability of the product during its shelf-life.
Predictive dissolution testing
Dissolution data can also be used as a prognostic tool to predict the behaviour and performance of oral solid drug products in the gastrointestinal tract. The aim is to correlate as closely as possible measured in-vitro parameters with oral bioavailability. This type of dissolution testing is known as predictive dissolution testing. Unlike QC dissolution testing, it may require dissolution test methods which reflect more closely the physiological makeup of the gastrointestinal tract. Such test conditions are not easy to devise, but if done appropriately, they will enable the development of in vitro/in vivo predictions.
These predictions can be simple qualitative or semi-quantitative relationships (IVIVR) or quantitative correlations established using mathematical models (IVIVC). In its simplest definition, an IVIVC is a correlation (preferably linear) between an in vitro feature of the drug product (in the present context, its dissolution characteristics) and a biological parameter (e.g. the uptake of the drug in vivo and subsequent drug blood levels). The establishment of such a correlation during predictive dissolution testing is one of the most important aspects of a dissolution test for a preparation undergoing formulation development.
Predictive dissolution data have two main applications. Firstly, they can guide the early development of a new drug product by selecting formulations that yield desired in-vivo dissolution characteristics. Secondly they serve as a surrogate for clinical studies. In order to introduce new generic drug products into the market, companies are required to demonstrate that the generic product yields statistically similar rates and extents of drug absorption to the innovator (or branded) drug product – in other words that the two products are bioequivalent. Bioequivalence has traditionally been demonstrated in appropriately designed clinical studies, which are both time-consuming and expensive. However, following the introduction of the Biopharmaceutics Classification System in 1995 (BCS, Chapter 21) it became apparent that the dissolution data of some drug products can be correlated to oral bioavailability. Since 2000, bioequivalence between immediate-release products of highly water-soluble and easily absorbed drugs can be established on the basis of dissolution data, provided that the excipients present in the formulation do not affect the absorption of the drug.
Dissolution testing
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


