FIGURE 404-4 Simplified approach to the differential diagnosis of diabetes insipidus. When symptoms suggest diabetes insipidus (DI), the syndrome should be differentiated from a genitourinary (GU) abnormality by measuring the 24-h urine volume and osmolarity on unrestricted fluid intake. If DI is confirmed, basal plasma arginine vasopressin (AVP) should be measured on unrestricted fluid intake. If AVP is normal or elevated (>1 pg/mL), the patient probably has nephrogenic DI. However, if plasma AVP is low or undetectable, the patient has either pituitary DI or primary polydipsia. In that case, magnetic resonance imaging (MRI) of the brain can be performed to differentiate between these two conditions by determining whether or not the normal posterior pituitary bright spot is visible on T1-weighted midsagittal images. In addition, the MRI anatomy of the pituitary hypothalamic area can be examined to look for evidence of pathology that sometimes causes pituitary DI or the dipsogenic form of primary polydipsia. MRI is not reliable for differential diagnosis unless nephrogenic DI has been excluded because the bright spot is also absent, small, or faint in this condition.
If MRI and/or AVP assays with the requisite sensitivity and specificity are unavailable and a fluid deprivation test is impractical or undesirable, a third way to differentiate between pituitary DI, nephrogenic DI, and primary polydipsia is a trial of desmopressin therapy. Such a trial should be conducted with very close monitoring of serum sodium as well as urine output, preferably in hospital, because desmopressin will produce hyponatremia in 8–24 h if the patient has primary polydipsia.
TREATMENT | DIABETES INSIPIDUS |
The signs and symptoms of uncomplicated pituitary DI can be eliminated by treatment with desmopressin (DDAVP), a synthetic analogue of AVP (Fig. 404-1). DDAVP acts selectively at V2 receptors to increase urine concentration and decrease urine flow in a dose-dependent manner. It is also more resistant to degradation than is AVP and has a three- to fourfold longer duration of action. DDAVP can be given by IV or SC injection, nasal inhalation, or orally by means of a tablet of melt. The doses required to control pituitary DI completely vary widely, depending on the patient and the route of administration. However, among adults, they usually range from 1–2 μg qd or bid by injection, 10–20 μg bid or tid by nasal spray, or 100–400 μg bid or tid orally. The onset of antidiuresis is rapid, ranging from as little as 15 min after injection to 60 min after oral administration. When given in a dose that normalizes 24-h urinary osmolarity (400–800 mosmol/L) and volume (15–30 mL/kg body weight), DDAVP produces a slight (1–3%) increase in total body water and a decrease in plasma osmolarity/sodium that rapidly eliminates thirst and polydipsia (Fig. 404-5). Consequently, water balance is maintained within the normal range. Hyponatremia does not develop unless urine volume is reduced too far (to less than 10 mL/kg per day) or fluid intake is excessive due to an associated abnormality in thirst or cognition. Fortunately, thirst abnormalities are rare, and if the patient is taught to drink only when truly thirsty, DDAVP can be given safely in doses sufficient to normalize urine output (~15–30 mL/kg per day) without the need for allowing intermittent escape to prevent water intoxication.
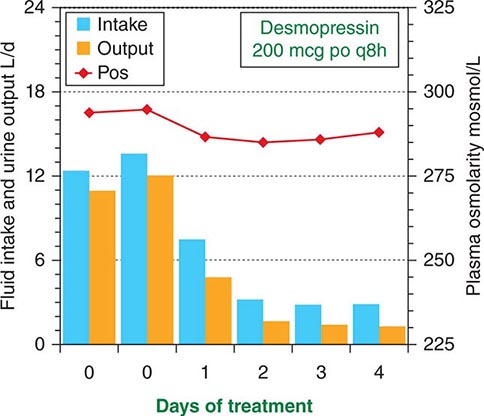
FIGURE 404-5 Effect of desmopressin therapy on fluid intake (blue bars), urine output (orange bars), and plasma osmolarity (red line) in a patient with uncomplicated pituitary diabetes insipidus. Note that treatment rapidly reduces fluid intake and urine output tonormal, with only a slight increase in body water as evidenced by the slight decrease in plasma osmolarity.
Primary polydipsia cannot be treated safely with DDAVP or any other antidiuretic drug because eliminating the polyuria does not eliminate the urge to drink. Therefore, it invariably produces hyponatremia and/or other signs of water intoxication, usually within 8–24 h if urine output is normalized completely. There is no consistently effective way to correct dipsogenic or psychogenic polydipsia, but the iatrogenic form may respond to patient education. To minimize the risk of water intoxication, all patients should be warned about the use of other drugs such as thiazide diuretics or carbamazepine (Tegretol) that can impair urinary free-water excretion directly or indirectly.
The polyuria and polydipsia of nephrogenic DI are not affected by treatment with standard doses of DDAVP. If resistance is partial, it may be overcome by tenfold higher doses, but this treatment is too expensive and inconvenient for long-term use. However, treatment with conventional doses of a thiazide diuretic and/or amiloride in conjunction with a low-sodium diet and a prostaglandin synthesis inhibitor (e.g., indomethacin) usually reduces the polyuria and polydipsia by 30–70% and may eliminate them completely in some patients. Side effects such as hypokalemia and gastric irritation can be minimized by the use of amiloride or potassium supplements and by taking medications with meals.
HYPODIPSIC HYPERNATREMIA
An increase in plasma osmolarity/sodium above the normal range (hypertonic hypernatremia) can be caused by either a decrease in total body water or an increase in total body sodium. The former results from a failure to drink enough to replace normal or increased urinary and insensible water loss. The deficient intake can be due either to water deprivation or a lack of thirst (hypodipsia). The most common cause of an increase in total body sodium is primary hyperaldosteronism (Chap. 406). Rarely, it can also result from ingestion of hypertonic saline in the form of sea water or incorrectly prepared infant formula. However, even in these forms of hypernatremia, inadequate intake of water also contributes. This chapter focuses on hypodipsic hypernatremia, the form of hypernatremia due to a primary defect in the thirst mechanism.
Clinical Characteristics Hypodipsic hypernatremia is a syndrome characterized by chronic or recurrent hypertonic dehydration. The hypernatremia varies widely in severity and usually is associated with signs of hypovolemia such as tachycardia, postural hypotension, azotemia, hyperuricemia, and hypokalemia due to secondary hyperaldosteronism. Muscle weakness, pain, rhabdomyolysis, hyperglycemia, hyperlipidemia, and acute renal failure may also occur. Obtundation or coma may be present but are often absent. Despite inappropriately low levels of plasma AVP, DI usually is not evident at presentation but may develop during rehydration as blood volume, blood pressure, and plasma osmolarity/sodium return toward normal, further reducing plasma AVP.
Etiology Hypodipsia is usually due to hypogenesis or destruction of the osmoreceptors in the anterior hypothalamus that regulate thirst. These defects can result from various congenital malformations of midline brain structures or may be acquired due to diseases such as occlusions of the anterior communicating artery, primary or metastatic tumors in the hypothalamus, head trauma, surgery, granulomatous diseases such as sarcoidosis and histiocytosis, AIDS, and cytomegalovirus encephalitis. Because of their proximity, the osmoreceptors that regulate AVP secretion also are usually impaired. Thus, AVP secretion responds poorly or not at all to hyperosmotic stimulation (Fig. 404-6) but, in most cases, increases normally to nonosmotic stimuli such as nausea or large reductions in blood volume or blood pressure, indicating that the neurohypophysis is intact.
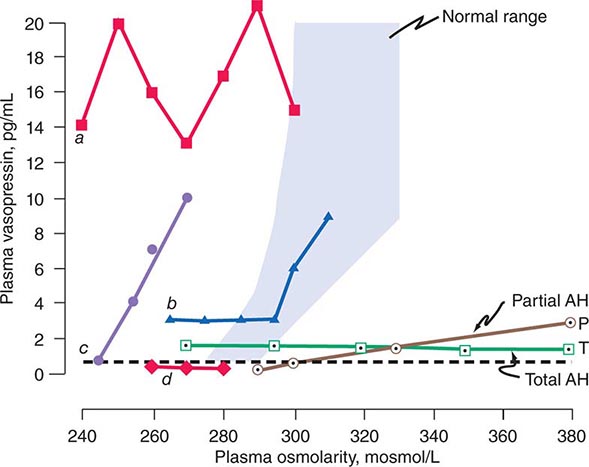
FIGURE 404-6 Heterogeneity of osmoregulatory dysfunction in adipsic hypernatremia (AH) and the syndrome of inappropriate antidiuresis (SIAD). Each line depicts schematically the relationship of plasma arginine vasopressin (AVP) to plasma osmolarity during water loading and/or infusion of 3% saline in a patient with either AH (open symbols) or SIAD (closed symbols). The shaded area indicates the normal range of the relationship. The horizontal broken line indicates the plasma AVP level below which the hormone is undetectable and urinary concentration usually does not occur. Lines P and T represent patients with a selective deficiency in the osmoregulation of thirst and AVP that is either partial (![]() ) or total (
) or total (![]() ). In the latter, plasma AVP does not change in response to increases or decreases in plasma osmolarity but remains within a range sufficient to concentrate the urine even if overhydration produces hypotonic hyponatremia. In contrast, if the osmoregulatory deficiency is partial (
). In the latter, plasma AVP does not change in response to increases or decreases in plasma osmolarity but remains within a range sufficient to concentrate the urine even if overhydration produces hypotonic hyponatremia. In contrast, if the osmoregulatory deficiency is partial (![]() ), rehydration of the patient suppresses plasma AVP to levels that result in urinary dilution and polyuria before plasma osmolarity and sodium are reduced to normal. Lines a –d represent different defects in the osmoregulation of plasma AVP observed in patients with SIADH or SIAD. In a (
), rehydration of the patient suppresses plasma AVP to levels that result in urinary dilution and polyuria before plasma osmolarity and sodium are reduced to normal. Lines a –d represent different defects in the osmoregulation of plasma AVP observed in patients with SIADH or SIAD. In a (![]() ), plasma AVP is markedly elevated and fluctuates widely without relation to changes in plasma osmolarity, indicating complete loss of osmoregulation. In b (
), plasma AVP is markedly elevated and fluctuates widely without relation to changes in plasma osmolarity, indicating complete loss of osmoregulation. In b (![]() ), plasma AVP remains fixed at a slightly elevated level until plasma osmolarity reaches the normal range, at which point it begins to rise appropriately, indicating a selective defect in the inhibitory component of the osmoregulatory mechanism. In c (
), plasma AVP remains fixed at a slightly elevated level until plasma osmolarity reaches the normal range, at which point it begins to rise appropriately, indicating a selective defect in the inhibitory component of the osmoregulatory mechanism. In c (![]() ), plasma AVP rises in close correlation with plasma osmolarity before the latter reaches the normal range, indicating downward resetting of the osmostat. In d (
), plasma AVP rises in close correlation with plasma osmolarity before the latter reaches the normal range, indicating downward resetting of the osmostat. In d (![]() ), plasma AVP appears to be osmoregulated normally, suggesting that the inappropriate antidiuresis is caused by some other abnormality.
), plasma AVP appears to be osmoregulated normally, suggesting that the inappropriate antidiuresis is caused by some other abnormality.
Pathophysiology Hypodipsia results in a failure to drink enough water to replenish obligatory renal and extrarenal losses. Consequently, plasma osmolarity and sodium rise often to extremely high levels before the disorder is recognized. In most cases, urinary loss of water contributes little, if any, to the dehydration because AVP continues to be secreted in the small amounts necessary to concentrate the urine. In some patients this appears to be due to hypovolemic stimulation and/or incomplete destruction of AVP osmoreceptors because plasma AVP declines and DI develops during rehydration (Fig. 404-6). In others, however, plasma AVP does not decline during rehydration even if they are overhydrated. Consequently, they develop a hyponatremic syndrome indistinguishable from inappropriate antidiuresis. This suggests that the AVP osmoreceptors normally provide inhibitory and stimulatory input to the neurohypophysis and the patients can no longer osmotically stimulate or suppress tonic secretion of the hormone because both inputs have been totally eliminated by the same pathology that destroyed the osmoregulation of thirst. In a few patients, the neurohypophysis is also destroyed, resulting in a combination of chronic pituitary DI and hypodipsia that is particularly difficult to manage.
Differential Diagnosis Hypodipsic hypernatremia usually can be distinguished from other causes of inadequate fluid intake (e.g., coma, paralysis, restraints, absence of fresh water) by the clinical history and setting. Previous episodes and/or denial of thirst and failure to drink spontaneously when the patient is conscious, unrestrained, and hypernatremic are virtually diagnostic. The hypernatremia caused by excessive retention or intake of sodium can be distinguished by the presence of thirst as well as the physical and laboratory signs of hypervolemia rather than hypovolemia.
TREATMENT | HYPODIPSIC HYPERNATREMIA |
Hypodipsic hypernatremia should be treated by administering water orally if the patient is alert and cooperative or by infusing hypotonic fluids (0.45% saline or 5% dextrose and water) if the patient is not. The amount of free water in liters required to correct the deficit (ΔFW) can be estimated from body weight in kg (BW) and the serum sodium concentration in mmol/L (SNa) by the formula ΔFW = 0.5BW × ([SNa – 140]/140). If serum glucose (SGlu) is elevated, the measured S Na should be corrected (SNa*) by the formula SNa* = SNa + ([SGlu – 90]/36). This amount plus an allowance for continuing insensible and urinary losses should be given over a 24- to 48-h period. Close monitoring of serum sodium as well as fluid intake and urinary output is essential because, depending on the extent of osmoreceptor deficiency, some patients will develop AVP-deficient DI, requiring DDAVP therapy to complete rehydration; others will develop hyponatremia and a syndrome of inappropriate antidiuresis (SIAD)-like picture if overhydrated. If hyperglycemia and/or hypokalemia are present, insulin and/or potassium supplements should be given with the expectation that both can be discontinued soon after rehydration is complete. Plasma urea/creatinine should be monitored closely for signs of acute renal failure caused by rhabdomyolysis, hypovolemia, and hypotension.
Once the patient has been rehydrated, an MRI of the brain and tests of anterior pituitary function should be performed to look for the cause and collateral defects in other hypothalamic functions. A long-term management plan to prevent or minimize recurrence of the fluid and electrolyte imbalance also should be developed. This should include a practical method to regulate fluid intake in accordance with variations in water balance as indicated by changes in body weight or serum sodium determined by home monitoring analyzers. Prescribing a constant fluid intake is ineffective and potentially dangerous because it does not take into account the large, uncontrolled variations in insensible loss that inevitably result from changes in ambient temperature and physical activity.
HYPONATREMIA DUE TO INAPPROPRIATE ANTIDIURESIS
A decrease in plasma osmolarity/sodium below the normal range (hypotonic hyponatremia) can be due to any of three different types of salt and water imbalance: (1) an increase in total body water that exceeds the increase in total body sodium (hypervolemic hyponatremia); (2) a decrease in body sodium greater than the decrease in body water (hypovolemic hyponatremia); or (3) an increase in body water with little or no change in body sodium (euvolemic hyponatremia) (Chap. 63). All three forms are associated with a failure to fully dilute the urine and mount a water diuresis in the face of hypotonic hyponatremia. The hypervolemic form typically occurs in disorders like severe congestive heart failure or cirrhosis. The hypovolemic form typically occurs in disorders such as severe diarrhea, diuretic abuse, or mineralocorticoid deficiency. Euvolemic hyponatremia, however, is due mainly to expansion of total body water caused by excessive intake in the face of a defect in urinary dilution. The impaired dilution is usually caused by a defect in the osmotic suppression of AVP that can have either of two causes. One is a nonhemodynamic stimulus such as nausea or a cortisol deficiency, which can be corrected quickly by treatment with antiemetics or cortisol. The other is a primary defect in osmoregulation caused by another disorder such as malignancy, stroke, or pneumonia that cannot be easily or quickly corrected. The latter is commonly known as the syndrome of inappropriate antidiuretic hormone (SIADH). Much less often, euvolemic hyponatremia can also result from AVP-independent activation of renal V2 receptors, a variant known as nephrogenic inappropriate antidiuresis or NSIAD. Both of the latter will be discussed in this chapter.
Clinical Characteristics Antidiuresis of any cause decreases the volume and increases the concentration of urine. If not accompanied by a commensurate reduction in fluid intake or an increase in insensible loss, the reduction in urine output results in excess water retention which expands and dilutes body fluids. If the hyponatremia develops gradually or has been present for more than a few days, it may be largely asymptomatic. However, if it develops acutely, it is usually accompanied by symptoms and signs of water intoxication that may include mild headache, confusion, anorexia, nausea, vomiting, coma, and convulsions. Severe acute hyponatremia may be lethal. Other clinical signs and symptoms vary greatly, depending on the type of hyponatremia. The hypervolemic form is characterized by generalized edema and other signs of marked volume expansion. The opposite is evident in the hypovolemic form. However, overt signs of volume expansion or contraction are absent in SIADH, SIAD, and other forms of euvolemic hyponatremia.
Etiology In SIADH, the inappropriate secretion of AVP can have many different causes. They include ectopic production of AVP by lung cancer or other neoplasms; eutopic release induced by various diseases or drugs; and exogenous administration of AVP, DDAVP, or large doses of oxytocin (Table 404-2). The ectopic forms result from abnormal expression of the AVP-NPII gene by primary or metastatic malignancies. The eutopic forms occur most often in patients with acute infections or strokes but have also been associated with many other neurologic diseases and injuries. The mechanisms by which these diseases interfere with osmotic suppression of AVP are not known. The defect in osmoregulation can take any of four distinct forms (Fig. 404-6). In one of the most common (reset osmostat), AVP secretion remains fully responsive to changes in plasma osmolarity/sodium, but the threshold, or set point, of the osmoregulatory system is abnormally low. These patients differ from those with the other types of SIADH in that they are able to maximally suppress plasma AVP and dilute their urine if their fluid intake is high enough to reduce their plasma osmolarity and/or sodium to the new set point. In most patients, SIADH is self-limited and remits spontaneously within 2–3 weeks, but about 10% of cases are chronic. Another, smaller subgroup (~10% of the total) has inappropriate antidiuresis without a demonstrable defect in the osmoregulation of plasma AVP (Fig. 404-6). In some of them, all young boys, the inappropriate antidiuresis has been traced to a constitutively activating mutation of the V2 receptor gene. This unusual variant may be referred to as familial nephrogenic SIAD (NSIAD) to distinguish it from other possible causes of the syndrome. The inappropriate antidiuresis in these patients appears to be permanent, although the hyponatremia is variable owing presumably to individual differences in fluid intake.
CAUSES OF SYNDROME OF INAPPROPRIATE ANTIDIURETIC HORMONE (SIADH) |
Pathophysiology Impaired osmotic suppression of antidiuresis results in excessive retention of water and dilution of body fluids only if water intake exceeds insensible and urinary losses. The excess intake is sometimes due to an associated defect in the osmoregulation of thirst (dipsogenic) but can also be psychogenic or iatrogenic, including excessive IV administration of hypotonic fluids. In SIADH and other forms of euvolemic hyponatremia, the decrease in plasma osmolarity/sodium and the increase in extracellular and intracellular volume are proportional to the amount of water retained. Thus, an increase in body water of 10% (~4 L in a 70-kg adult) reduces plasma osmolarity and sodium by approximately 10% (~28 mosmol/L or 14 meq/L). An increase in body water of this magnitude is rarely detectable on physical examination but will be reflected in a weight gain of about 4 kg. It also increases glomerular filtration and atrial natriuretic hormone and suppresses plasma renin activity, thereby increasing urinary sodium excretion. The resultant reduction in total body sodium decreases the expansion of extracellular volume but aggravates the hyponatremia and further expands intracellular volume. The latter further increases brain swelling and intracranial pressure, which probably produces most of the symptoms of acute water intoxication. Within a few days, this swelling may be counteracted by inactivation or elimination of intracellular solutes, resulting in the remission of symptoms even though the hyponatremia persists.
In type I (hypervolemic) or type II (hypovolemic) hyponatremia, osmotic suppression of AVP secretion appears to be counteracted by a hemodynamic stimulus resulting from a large reduction in cardiac output and/or effective blood volume. The resultant antidiuresis is enhanced by decreased distal delivery of glomerular filtrate that results from increased reabsorption of sodium in proximal nephron. If the reduction in urine output is not associated with a commensurate reduction in water intake or an increase in insensible loss, body fluids are expanded and diluted, resulting in hyponatremia despite an increase in body sodium. Unlike SIADH and other forms of euvolemic hyponatremia, however, glomerular filtration is reduced and plasma renin activity and aldosterone are elevated. Thus, the rate of urinary sodium excretion is low (unless sodium reabsorption is impaired by a diuretic), and the hyponatremia is usually accompanied by edema, hypokalemia, azotemia, and hyperuricemia. In type II (hypovolemic) hyponatremia, sodium and water are also retained as an appropriate compensatory response to the severe depletion.
Differential Diagnosis SIADH is a diagnosis of exclusion that usually can be made from the history, physical examination, and basic laboratory data. If hyperglycemia is present, its contribution to the reduction in plasma sodium can be estimated either by measuring plasma osmolarity for a more accurate estimate of the true “effective” tonicity of body fluids or by correcting the measured plasma sodium for the reduction caused by the hyperglycemia using the simplified formula
![]()
where Pna = plasma sodium in meq/L and Pglu = plasma glucose in mg/dL.
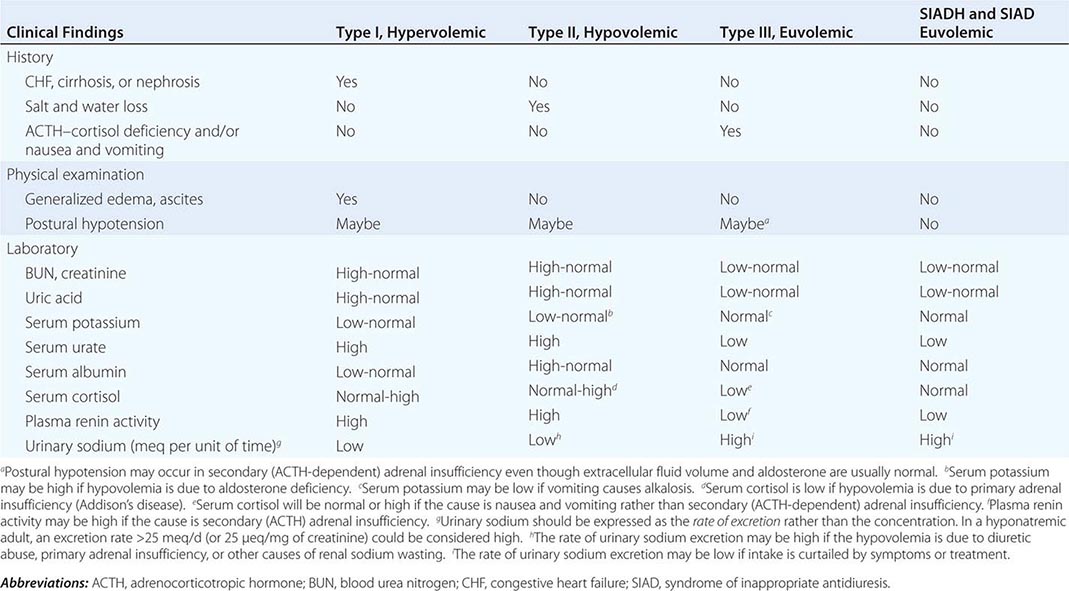
If the plasma osmolarity and/or corrected plasma sodium are below normal limits, hypotonic hyponatremia is present and further evaluation to determine the type should be undertaken in order to administer safe and effective treatment. This differentiation is usually possible by evaluating standard clinical indicators of the extracellular fluid volume (Table 404-3). If these findings are ambiguous or contradictory, measuring plasma renin activity or the rate of urinary sodium excretion may be helpful provided that the hyponatremia is not in the recovery phase or is due to a primary defect in renal conservation of sodium, diuretic abuse, or hyporeninemic hypoaldosteronism. The latter may be suspected if serum potassium is elevated instead of low, as it usually is in types I and II hyponatremia. Measurements of plasma AVP are currently of no value in differentiating SIADH from the other types of hyponatremia since the plasma levels are elevated similarly in all. In patients who fulfill the clinical criteria for type III (euvolemic) hyponatremia, morning plasma cortisol should also be measured to exclude secondary adrenal insufficiency. If it is normal and there is no history of nausea/vomiting, the diagnosis of SIADH is confirmed, and a careful search for occult lung cancer or other common causes of the syndrome (Table 404-2) should be undertaken.
DIFFERENTIAL DIAGNOSIS OF HYPONATREMIA BASED ON CLINICAL ASSESSMENT OF EXTRACELLULAR FLUID VOLUME (ECFV) |
SIAD due to an activating mutation of the V2 receptor gene should be suspected if the hyponatremia occurs in a child or several members of the family or is refractory to treatment with a vaptan (see below). In that case, plasma AVP should be measured to confirm that it is appropriately suppressed while the hyponatremia and antidiuresis are present, and the V2 receptor gene should be sequenced, if possible.
TREATMENT | HYPONATREMIA |
The management of hyponatremia differs depending on the type and the severity and duration of symptoms. In acute symptomatic SIADH, the aim should be to raise plasma osmolarity and/or plasma sodium at a rate approximating 1% an hour until they reach levels of about 270 mosmol/L or 130 meq/L, respectively. This can be accomplished in either of two ways. One is to infuse hypertonic (3%) saline at a rate of about 0.05 mL/kg body weight per minute. This treatment also has the advantage of correcting the sodium deficiency that is partly responsible for the hyponatremia and often produces a solute diuresis that serves to remove some of the excess water. The other treatment is to reduce body water by giving an AVP receptor-2 antagonist (vaptan) to block the antidiuretic effect of AVP and increase urine output (Fig. 404-7). One of the vaptans, a combined V2/V1a antagonist (Conivaptan), has been approved for short-term, in-hospital IV treatment of SIADH, and others are in various stages of development. With either approach, fluid intake should be restricted to less than urine output, and serum sodium should be checked at least once every 2h to ensure it is not raised too fast or too far. Doing so may result in central pontine myelinolysis, an acute, potentially fatal neurologic syndrome characterized by quadriparesis, ataxia, and abnormal extraocular movements.
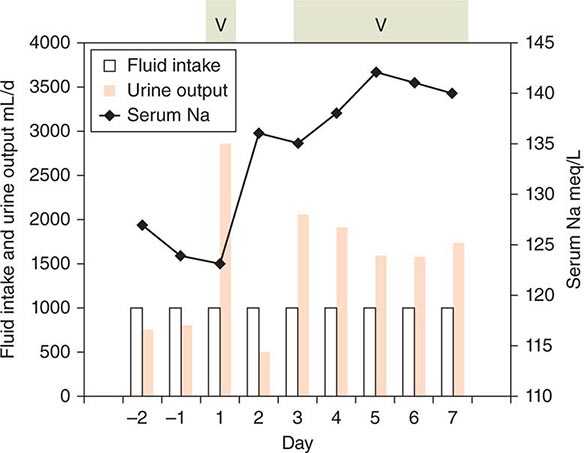
FIGURE 404-7 The effect of vaptan therapy on water balance in a patient with chronic syndrome of inappropriate antidiuretic hormone (SIADH). The periods of vaptan (V) therapy are indicated by the green shaded boxes at the top. Urine output is indicated by orange bars. Fluid intake is shown by the open bars. Intake was restricted to 1 L/d throughout. Serum sodium is indicated by the black line. Note that sodium increased progressively when vaptan increased urine output to levels that clearly exceeded fluid intake.
In chronic and/or minimally symptomatic SIADH, the hyponatremia can and should be corrected more gradually. This can be achieved by restricting total fluid intake to less than the sum of urinary and insensible losses. Because the water derived from food (300–700 mL/d) usually approximates basal insensible losses in adults, the aim should be to reduce total discretionary intake (all liquids) to approximately 500 mL less than urinary output. Adherence to this regimen is often problematic and, even if achieved, usually reduces body water and increases serum sodium by only about 1–2% per day. Hence, additional approaches are usually desirable if not necessary. The best approach for treatment of chronic SIADH is the administration of an oral vaptan, tolvaptan, a selective V2 antagonist that also increases urinary water excretion by blocking the antidiuretic effect of AVP. Some restriction of fluid intake may also be necessary to achieve satisfactory control of the hyponatremia. It is approved for treatment of nonemergent SIADH with initial in-hospital dosing. Other approaches include demeclocycline, 150–300 mg PO tid or qid, or fludrocortisone, 0.05–0.2 mg PO bid. The effect of the demeclocycline manifests in 7–14 days and is due to induction of a reversible form of nephrogenic DI. Potential side effects include phototoxicity and azotemia. The effect of fludrocortisone also requires 1–2 weeks and is partly due to increased retention of sodium and possibly inhibition of thirst. It also increases urinary potassium excretion, which may require replacement through dietary adjustments or supplements and may induce hypertension, occasionally necessitating discontinuation of the treatment.
In euvolemic hyponatremia caused by protracted nausea and vomiting or isolated glucocorticoid deficiency (type III), all abnormalities can be corrected quickly and completely by giving an antiemetic or stress doses of hydrocortisone (for glucocorticoid deficiency). As with other treatments, care must be taken to ensure that serum sodium does not rise too quickly or too far.
In SIAD due to an activating mutation of the V2 receptor, the V2 antagonists usually do not block the antidiuresis or raise plasma osmolarity/sodium. In that condition, use of an osmotic diuretic such as urea is reported to be effective in preventing or correcting hyponatremia. However, some vaptans may be effective in patients with a different type of activating mutation so the response to this therapy may be neither predictable nor diagnostic.
In hypervolemic hyponatremia, fluid restriction is also appropriate and somewhat effective if it can be maintained. However, infusion of hypertonic saline is contraindicated because it further increases total body sodium and edema and may precipitate cardiovascular decompensation. However, as in SIADH, the V2 receptor antagonists are also safe and effective in the treatment of hypervolemic hyponatremia caused by congestive heart failure. Tolvaptan is approved by the Food and Drug Administration for this indication with the caveat that treatment should be initiated or reinitiated in hospital. Its use should also be limited to 30 days at a time because of reports that longer periods may be associated with abnormal liver chemistries.
In hypovolemic hyponatremia, the defect in AVP secretion and water balance usually can be corrected easily and quickly by stopping the loss of sodium and water and/or replacing the deficits by mouth or IV infusion of normal or hypertonic saline. As with the treatment of other forms of hyponatremia, care must be taken to ensure that plasma sodium does not increase too rapidly or too far. Fluid restriction and administration of AVP antagonists are contraindicated in type II hyponatremia because they would only aggravate the underlying volume depletion and could result in hemodynamic collapse.
GLOBAL PERSPECTIVES
 The incidence, clinical characteristics, etiology, pathophysiology, differential diagnosis, and treatments of fluid and electrolyte disorders in tropical and nonindustrialized countries differ in some respects from those in the United States and other industrialized parts of the world. Hyponatremia, for example, appears to be more common and is more likely to be due to infectious diseases such as cholera, shigellosis, and other diarrheal disorders. In these circumstances, hyponatremia is probably due to gastrointestinal losses of salt and water (hypovolemia type II), but other abnormalities, including undefined infectious toxins, also may contribute. The causes of DI are similar worldwide except that malaria and venoms from snake or insect bites are much more common.
The incidence, clinical characteristics, etiology, pathophysiology, differential diagnosis, and treatments of fluid and electrolyte disorders in tropical and nonindustrialized countries differ in some respects from those in the United States and other industrialized parts of the world. Hyponatremia, for example, appears to be more common and is more likely to be due to infectious diseases such as cholera, shigellosis, and other diarrheal disorders. In these circumstances, hyponatremia is probably due to gastrointestinal losses of salt and water (hypovolemia type II), but other abnormalities, including undefined infectious toxins, also may contribute. The causes of DI are similar worldwide except that malaria and venoms from snake or insect bites are much more common.
405 | Disorders of the Thyroid Gland |
The thyroid gland produces two related hormones, thyroxine (T4) and triiodothyronine (T3) (Fig. 405-1). Acting through thyroid hormone receptors α and β, these hormones play a critical role in cell differentiation during development and help maintain thermogenic and metabolic homeostasis in the adult. Autoimmune disorders of the thyroid gland can stimulate overproduction of thyroid hormones (thyrotoxicosis) or cause glandular destruction and hormone deficiency (hypothyroidism). In addition, benign nodules and various forms of thyroid cancer are relatively common and amenable to detection by physical examination.
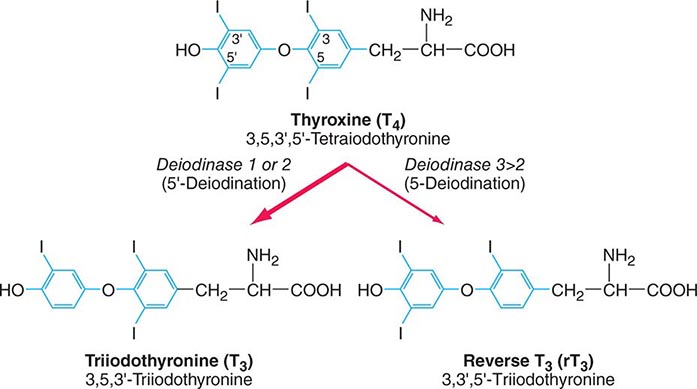
FIGURE 405-1 Structures of thyroid hormones. Thyroxine (T4) contains four iodine atoms. Deiodination leads to production of the potent hormone triiodothyronine (T3) or the inactive hormone reverse T3.
ANATOMY AND DEVELOPMENT
The thyroid (Greek thyreos, shield, plus eidos, form) consists of two lobes connected by an isthmus. It is located anterior to the trachea between the cricoid cartilage and the suprasternal notch. The normal thyroid is 12–20 g in size, highly vascular, and soft in consistency. Four parathyroid glands, which produce parathyroid hormone (Chap. 424), are located posterior to each pole of the thyroid. The recurrent laryngeal nerves traverse the lateral borders of the thyroid gland and must be identified during thyroid surgery to avoid injury and vocal cord paralysis.
The thyroid gland develops from the floor of the primitive pharynx during the third week of gestation. The developing gland migrates along the thyroglossal duct to reach its final location in the neck. This feature accounts for the rare ectopic location of thyroid tissue at the base of the tongue (lingual thyroid) as well as the occurrence of thyroglossal duct cysts along this developmental tract. Thyroid hormone synthesis normally begins at about 11 weeks’ gestation.
Neural crest derivatives from the ultimobranchial body give rise to thyroid medullary C cells that produce calcitonin, a calcium-lowering hormone. The C cells are interspersed throughout the thyroid gland, although their density is greatest in the juncture of the upper one-third and lower two-thirds of the gland. Calcitonin plays a minimal role in calcium homeostasis in humans but the C-cells are important because of their involvement in medullary thyroid cancer.
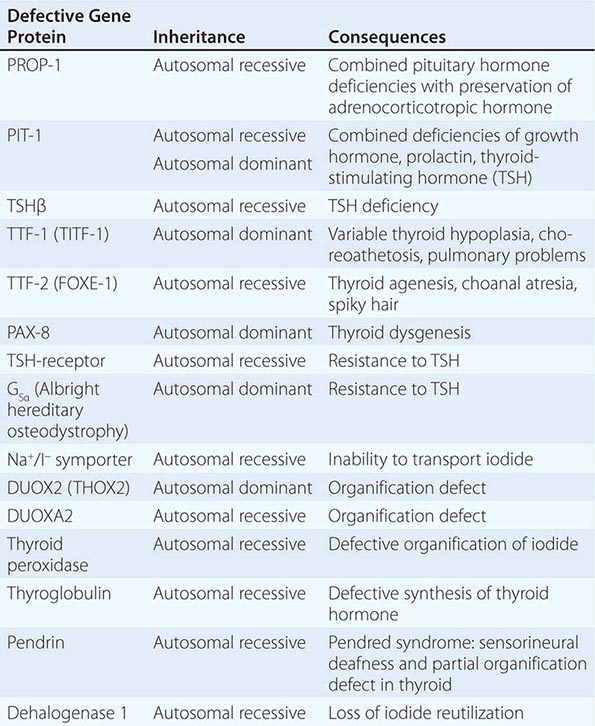
Thyroid gland development is orchestrated by the coordinated expression of several developmental transcription factors. Thyroid transcription factor (TTF)-1, TTF-2, and paired homeobox-8 (PAX-8) are expressed selectively, but not exclusively, in the thyroid gland. In combination, they dictate thyroid cell development and the induction of thyroid-specific genes such as thyroglobulin (Tg), thyroid peroxidase (TPO), the sodium iodide symporter (Na+/I–, NIS), and the thyroid-stimulating hormone receptor (TSH-R). Mutations in these developmental transcription factors or their downstream target genes are rare causes of thyroid agenesis or dyshormonogenesis, although the causes of most forms of congenital hypothyroidism remain unknown (Table 405-1). Because congenital hypothyroidism occurs in approximately 1 in 4000 newborns, neonatal screening is now performed in most industrialized countries (see below). Transplacental passage of maternal thyroid hormone occurs before the fetal thyroid gland begins to function and provides partial hormone support to a fetus with congenital hypothyroidism. Early thyroid hormone replacement in newborns with congenital hypothyroidism prevents potentially severe developmental abnormalities.
GENETIC CAUSES OF CONGENITAL HYPOTHYROIDISM |
The thyroid gland consists of numerous spherical follicles composed of thyroid follicular cells that surround secreted colloid, a proteinaceous fluid containing large amounts of thyroglobulin, the protein precursor of thyroid hormones (Fig. 405-2). The thyroid follicular cells are polarized—the basolateral surface is apposed to the bloodstream and an apical surface faces the follicular lumen. Increased demand for thyroid hormone is regulated by thyroid-stimulating hormone (TSH), which binds to its receptor on the basolateral surface of the follicular cells. This binding leads to Tg reabsorption from the follicular lumen and proteolysis within the cytoplasm, yielding thyroid hormones for secretion into the bloodstream.
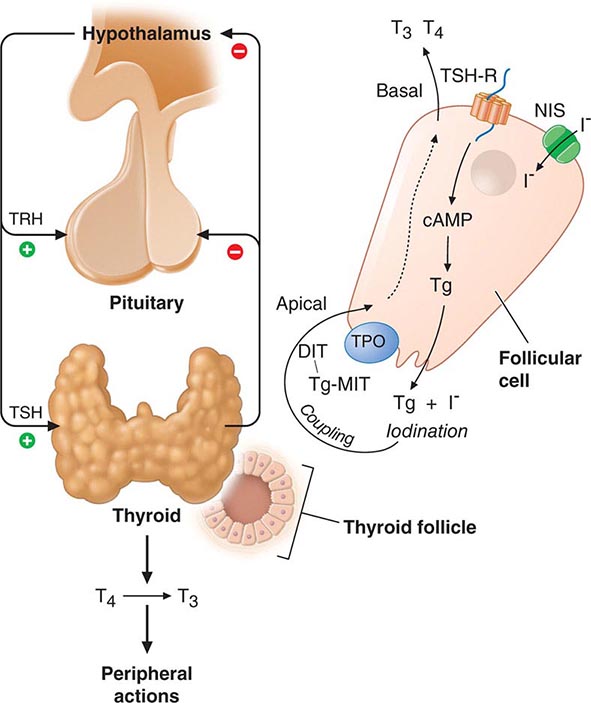
FIGURE 405-2 Regulation of thyroid hormone synthesis. Left. Thyroid hormones T4 and T3 feed back to inhibit hypothalamic production of thyrotropin-releasing hormone (TRH) and pituitary production of thyroid-stimulating hormone (TSH). TSH stimulates thyroid gland production of T4 and T3. Right. Thyroid follicles are formed by thyroid epithelial cells surrounding proteinaceous colloid, which contains thyroglobulin. Follicular cells, which are polarized, synthesize thyroglobulin and carry out thyroid hormone biosynthesis (see text for details). DIT, diiodotyrosine; MIT, monoiodotyrosine; NIS, sodium iodide symporter; Tg, thyroglobulin; TPO, thyroid peroxidase; TSH-R, thyroid-stimulating hormone receptor.
REGULATION OF THE THYROID AXIS
TSH, secreted by the thyrotrope cells of the anterior pituitary, plays a pivotal role in control of the thyroid axis and serves as the most useful physiologic marker of thyroid hormone action. TSH is a 31-kDa hormone composed of α and β subunits; the α subunit is common to the other glycoprotein hormones (luteinizing hormone, follicle-stimulating hormone, human chorionic gonadotropin [hCG]), whereas the TSH β subunit is unique to TSH. The extent and nature of carbohydrate modification are modulated by thyrotropin-releasing hormone (TRH) stimulation and influence the biologic activity of the hormone.
The thyroid axis is a classic example of an endocrine feedback loop. Hypothalamic TRH stimulates pituitary production of TSH, which, in turn, stimulates thyroid hormone synthesis and secretion. Thyroid hormones act via negative feedback predominantly through thyroid hormone receptor β2 (TRβ2) to inhibit TRH and TSH production (Fig. 405-2). The “set-point” in this axis is established by TSH. TRH is the major positive regulator of TSH synthesis and secretion. Peak TSH secretion occurs ~15 min after administration of exogenous TRH. Dopamine, glucocorticoids, and somatostatin suppress TSH but are not of major physiologic importance except when these agents are administered in pharmacologic doses. Reduced levels of thyroid hormone increase basal TSH production and enhance TRH-mediated stimulation of TSH. High thyroid hormone levels rapidly and directly suppress TSH gene expression secretion and inhibit TRH stimulation of TSH, indicating that thyroid hormones are the dominant regulator of TSH production. Like other pituitary hormones, TSH is released in a pulsatile manner and exhibits a diurnal rhythm; its highest levels occur at night. However, these TSH excursions are modest in comparison to those of other pituitary hormones, in part, because TSH has a relatively long plasma half-life (50 min). Consequently, single measurements of TSH are adequate for assessing its circulating level. TSH is measured using immunoradiometric assays that are highly sensitive and specific. These assays readily distinguish between normal and suppressed TSH values; thus, TSH can be used for the diagnosis of hyperthyroidism (low TSH) as well as hypothyroidism (high TSH).
THYROID HORMONE SYNTHESIS, METABOLISM, AND ACTION
THYROID HORMONE SYNTHESIS
Thyroid hormones are derived from Tg, a large iodinated glycoprotein. After secretion into the thyroid follicle, Tg is iodinated on tyrosine residues that are subsequently coupled via an ether linkage. Reuptake of Tg into the thyroid follicular cell allows proteolysis and the release of newly synthesized T4 and T3.
Iodine Metabolism and Transport Iodide uptake is a critical first step in thyroid hormone synthesis. Ingested iodine is bound to serum proteins, particularly albumin. Unbound iodine is excreted in the urine. The thyroid gland extracts iodine from the circulation in a highly efficient manner. For example, 10–25% of radioactive tracer (e.g., 123I) is taken up by the normal thyroid gland over 24 h; this value can rise to 70–90% in Graves’ disease. Iodide uptake is mediated by NIS, which is expressed at the basolateral membrane of thyroid follicular cells. NIS is most highly expressed in the thyroid gland, but low levels are present in the salivary glands, lactating breast, and placenta. The iodide transport mechanism is highly regulated, allowing adaptation to variations in dietary supply. Low iodine levels increase the amount of NIS and stimulate uptake, whereas high iodine levels suppress NIS expression and uptake. The selective expression of NIS in the thyroid allows isotopic scanning, treatment of hyperthyroidism, and ablation of thyroid cancer with radioisotopes of iodine, without significant effects on other organs. Mutation of the NIS gene is a rare cause of congenital hypothyroidism, underscoring its importance in thyroid hormone synthesis. Another iodine transporter, pendrin, is located on the apical surface of thyroid cells and mediates iodine efflux into the lumen. Mutation of the pendrin gene causes Pendred syndrome, a disorder characterized by defective organification of iodine, goiter, and sensorineural deafness.
 Iodine deficiency is prevalent in many mountainous regions and in central Africa, central South America, and northern Asia (Fig. 405-3). Europe remains mildly iodine-deficient, and health surveys indicate that iodine intake has been falling in the United States and Australia. The World Health Organization (WHO) estimates that about 2 billion people are iodine-deficient, based on urinary excretion data. In areas of relative iodine deficiency, there is an increased prevalence of goiter and, when deficiency is severe, hypothyroidism and cretinism. Cretinism is characterized by mental and growth retardation and occurs when children who live in iodine-deficient regions are not treated with iodine or thyroid hormone to restore normal thyroid hormone levels during early life. These children are often born to mothers with iodine deficiency, and it is likely that maternal thyroid hormone deficiency worsens the condition. Concomitant selenium deficiency may also contribute to the neurologic manifestations of cretinism. Iodine supplementation of salt, bread, and other food substances has markedly reduced the prevalence of cretinism. Unfortunately, however, iodine deficiency remains the most common cause of preventable mental deficiency, often because of societal resistance to food additives or the cost of supplementation. In addition to overt cretinism, mild iodine deficiency can lead to subtle reduction of IQ. Oversupply of iodine, through supplements or foods enriched in iodine (e.g., shellfish, kelp), is associated with an increased incidence of autoimmune thyroid disease. The recommended average daily intake of iodine is 150–250 μg/d for adults, 90–120 μg/d for children, and 250 μg/d for pregnant and lactating women. Urinary iodine is >10 μg/dL in iodine-sufficient populations.
Iodine deficiency is prevalent in many mountainous regions and in central Africa, central South America, and northern Asia (Fig. 405-3). Europe remains mildly iodine-deficient, and health surveys indicate that iodine intake has been falling in the United States and Australia. The World Health Organization (WHO) estimates that about 2 billion people are iodine-deficient, based on urinary excretion data. In areas of relative iodine deficiency, there is an increased prevalence of goiter and, when deficiency is severe, hypothyroidism and cretinism. Cretinism is characterized by mental and growth retardation and occurs when children who live in iodine-deficient regions are not treated with iodine or thyroid hormone to restore normal thyroid hormone levels during early life. These children are often born to mothers with iodine deficiency, and it is likely that maternal thyroid hormone deficiency worsens the condition. Concomitant selenium deficiency may also contribute to the neurologic manifestations of cretinism. Iodine supplementation of salt, bread, and other food substances has markedly reduced the prevalence of cretinism. Unfortunately, however, iodine deficiency remains the most common cause of preventable mental deficiency, often because of societal resistance to food additives or the cost of supplementation. In addition to overt cretinism, mild iodine deficiency can lead to subtle reduction of IQ. Oversupply of iodine, through supplements or foods enriched in iodine (e.g., shellfish, kelp), is associated with an increased incidence of autoimmune thyroid disease. The recommended average daily intake of iodine is 150–250 μg/d for adults, 90–120 μg/d for children, and 250 μg/d for pregnant and lactating women. Urinary iodine is >10 μg/dL in iodine-sufficient populations.
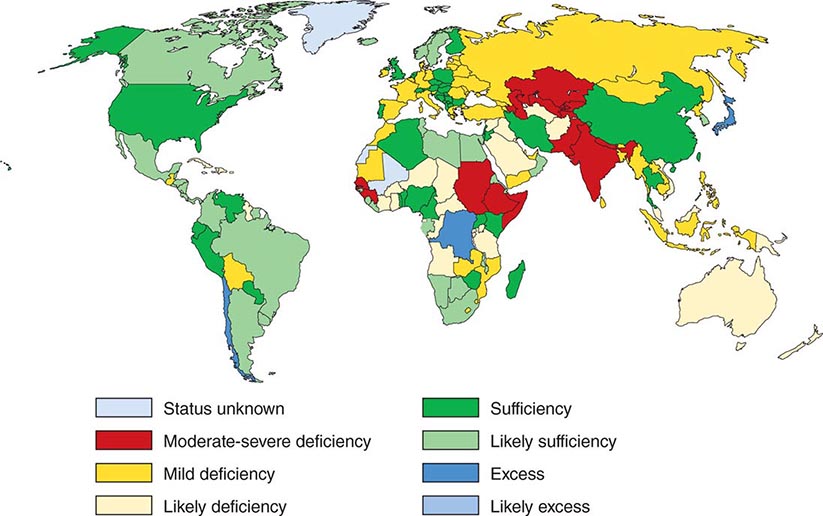
FIGURE 405-3 Worldwide iodine nutrition. Data are from the World Health Organization and the International Council for the Control of Iodine Deficiency Disorders (http://indorgs.virginia.edu/iccidd/mi/cidds.html).
Organification, Coupling, Storage, and Release After iodide enters the thyroid, it is trapped and transported to the apical membrane of thyroid follicular cells, where it is oxidized in an organification reaction that involves TPO and hydrogen peroxide produced by dual oxidase (DUOX) and DUOX maturation factor (DUOXA). The reactive iodine atom is added to selected tyrosyl residues within Tg, a large (660 kDa) dimeric protein that consists of 2769 amino acids. The iodotyrosines in Tg are then coupled via an ether linkage in a reaction that is also catalyzed by TPO. Either T4 or T3 can be produced by this reaction, depending on the number of iodine atoms present in the iodotyrosines. After coupling, Tg is taken back into the thyroid cell, where it is processed in lysosomes to release T4 and T3. Uncoupled mono- and diiodotyrosines (MIT, DIT) are deiodinated by the enzyme dehalogenase, thereby recycling any iodide that is not converted into thyroid hormones.
Disorders of thyroid hormone synthesis are rare causes of congenital hypothyroidism. The vast majority of these disorders are due to recessive mutations in TPO or Tg, but defects have also been identified in the TSH-R, NIS, pendrin, hydrogen peroxide generation, and dehalogenase. Because of the biosynthetic defect, the gland is incapable of synthesizing adequate amounts of hormone, leading to increased TSH and a large goiter.
TSH Action TSH regulates thyroid gland function through the TSH-R, a seven-transmembrane G protein–coupled receptor (GPCR). The TSH-R is coupled to the α subunit of stimulatory G protein (GSα), which activates adenylyl cyclase, leading to increased production of cyclic adenosine monophosphate (AMP). TSH also stimulates phosphatidylinositol turnover by activating phospholipase C. The functional role of the TSH-R is exemplified by the consequences of naturally occurring mutations. Recessive loss-of-function mutations cause thyroid hypoplasia and congenital hypothyroidism. Dominant gain-of-function mutations cause sporadic or familial hyperthyroidism that is characterized by goiter, thyroid cell hyperplasia, and autonomous function. Most of these activating mutations occur in the transmembrane domain of the receptor. They mimic the conformational changes induced by TSH binding or the interactions of thyroid-stimulating immunoglobulins (TSI) in Graves’ disease. Activating TSH-R mutations also occur as somatic events, leading to clonal selection and expansion of the affected thyroid follicular cell and autonomously functioning thyroid nodules (see below).
Other Factors That Influence Hormone Synthesis and Release Although TSH is the dominant hormonal regulator of thyroid gland growth and function, a variety of growth factors, most produced locally in the thyroid gland, also influence thyroid hormone synthesis. These include insulin-like growth factor I (IGF-I), epidermal growth factor, transforming growth factor β (TGF-β), endothelins, and various cytokines. The quantitative roles of these factors are not well understood, but they are important in selected disease states. In acromegaly, for example, increased levels of growth hormone and IGF-I are associated with goiter and predisposition to multinodular goiter (MNG). Certain cytokines and interleukins (ILs) produced in association with autoimmune thyroid disease induce thyroid growth, whereas others lead to apoptosis. Iodine deficiency increases thyroid blood flow and upregulates the NIS, stimulating more efficient iodine uptake. Excess iodide transiently inhibits thyroid iodide organification, a phenomenon known as the Wolff-Chaikoff effect. In individuals with a normal thyroid, the gland escapes from this inhibitory effect and iodide organification resumes; the suppressive action of high iodide may persist, however, in patients with underlying autoimmune thyroid disease.
THYROID HORMONE TRANSPORT AND METABOLISM
Serum Binding Proteins T4 is secreted from the thyroid gland in about twentyfold excess over T3 (Table 405-2). Both hormones are bound to plasma proteins, including thyroxine-binding globulin (TBG), transthyretin (TTR, formerly known as thyroxine-binding prealbumin, or TBPA), and albumin. The plasma-binding proteins increase the pool of circulating hormone, delay hormone clearance, and may modulate hormone delivery to selected tissue sites. The concentration of TBG is relatively low (1–2 mg/dL), but because of its high affinity for thyroid hormones (T4 > T3), it carries about 80% of the bound hormones. Albumin has relatively low affinity for thyroid hormones but has a high plasma concentration (~3.5 g/dL), and it binds up to 10% of T4 and 30% of T3. TTR carries about 10% of T4 but little T3.
CHARACTERISTICS OF CIRCULATING T4 AND T3 |
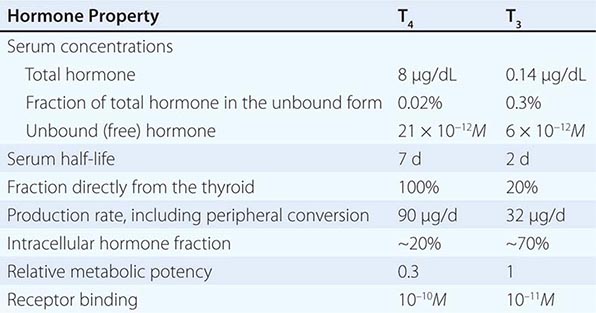
When the effects of the various binding proteins are combined, approximately 99.98% of T4 and 99.7% of T3 are protein-bound. Because T3 is less tightly bound than T4, the fraction of unbound T3 is greater than unbound T4, but there is less unbound T3 in the circulation because it is produced in smaller amounts and cleared more rapidly than T4. The unbound or “free” concentrations of the hormones are ~2 × 10–11 M for T4 and ~6 × 10–12 M for T3, which roughly correspond to the thyroid hormone receptor binding constants for these hormones (see below). The unbound hormone is thought to be biologically available to tissues. Nonetheless, the homeostatic mechanisms that regulate the thyroid axis are directed toward maintenance of normal concentrations of unbound hormones.
Abnormalities of Thyroid Hormone Binding Proteins A number of inherited and acquired abnormalities affect thyroid hormone binding proteins. X-linked TBG deficiency is associated with very low levels of total T4 and T3. However, because unbound hormone levels are normal, patients are euthyroid and TSH levels are normal. It is important to recognize this disorder to avoid efforts to normalize total T4 levels, because this leads to thyrotoxicosis and is futile because of rapid hormone clearance in the absence of TBG. TBG levels are elevated by estrogen, which increases sialylation and delays TBG clearance. Consequently, in women who are pregnant or taking estrogen-containing contraceptives, elevated TBG increases total T4 and T3 levels; however, unbound T4 and T3 levels are normal. These features are part of the explanation for why women with hypothyroidism require increased amounts of L-thyroxine replacement as TBG levels are increased by pregnancy or estrogen treatment. Mutations in TBG, TTR, and albumin may increase the binding affinity for T4 and/or T3 and cause disorders known as euthyroid hyperthyroxinemia or familial dysalbuminemic hyperthyroxinemia (FDH) (Table 405-3). These disorders result in increased total T4 and/or T3, but unbound hormone levels are normal. The familial nature of the disorders, and the fact that TSH levels are normal rather than suppressed, should suggest this diagnosis. Unbound hormone levels (ideally measured by dialysis) are normal in FDH. The diagnosis can be confirmed by using tests that measure the affinities of radiolabeled hormone binding to specific transport proteins or by performing DNA sequence analyses of the abnormal transport protein genes.
CONDITIONS ASSOCIATED WITH EUTHYROID HYPERTHYROXINEMIA |
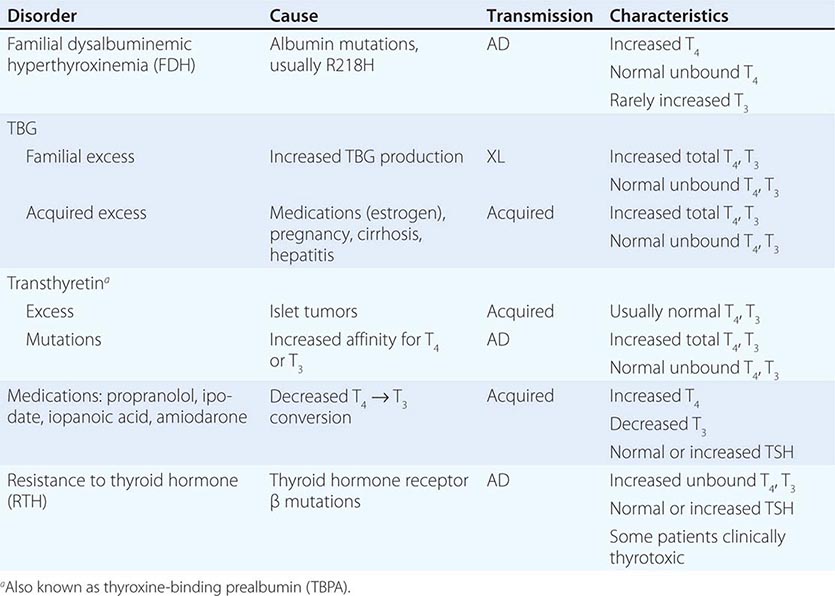
Abbreviations: AD, autosomal dominant; TBG, thyroxine-binding globulin; TSH, thyroid-stimulating hormone; XL, X-linked.
Certain medications, such as salicylates and salsalate, can displace thyroid hormones from circulating binding proteins. Although these drugs transiently perturb the thyroid axis by increasing free thyroid hormone levels, TSH is suppressed until a new steady state is reached, thereby restoring euthyroidism. Circulating factors associated with acute illness may also displace thyroid hormone from binding proteins (see “Sick Euthyroid Syndrome,” below).
Deiodinases T4 may be thought of as a precursor for the more potent T3. T4 is converted to T3 by the deiodinase enzymes (Fig. 405-1). Type I deiodinase, which is located primarily in thyroid, liver, and kidneys, has a relatively low affinity for T4. Type II deiodinase has a higher affinity for T4 and is found primarily in the pituitary gland, brain, brown fat, and thyroid gland. Expression of type II deiodinase allows it to regulate T3 concentrations locally, a property that may be important in the context of levothyroxine (T4) replacement. Type II deiodinase is also regulated by thyroid hormone; hypothyroidism induces the enzyme, resulting in enhanced T4 → T3 conversion in tissues such as brain and pituitary. T4 → T3 conversion is impaired by fasting, systemic illness or acute trauma, oral contrast agents, and a variety of medications (e.g., propylthiouracil, propranolol, amiodarone, glucocorticoids). Type III deiodinase inactivates T4 and T3 and is the most important source of reverse T3 (rT3), including in the sick euthyroid syndrome. This enzyme is expressed in the human placenta but is not active in healthy individuals. In the sick euthyroid syndrome, especially with hypoperfusion, the type III deiodinase is activated in muscle and liver. Massive hemangiomas that express type III deiodinase are a rare cause of hypothyroidism in infants.
THYROID HORMONE ACTION
Thyroid Hormone Transport Circulating thyroid hormones enter cells by passive diffusion and via specific transporters such as the monocarboxylate 8 transporter (MCT8), MCT10, and organic anion-transporting polypeptide 1C1. Mutations in the MCT8 gene have been identified in patients with X-linked psychomotor retardation and thyroid function abnormalities (low T4, high T3, and high TSH). After entering cells, thyroid hormones act primarily through nuclear receptors, although they also have nongenomic actions through stimulating mitochondrial enzymatic responses and may act directly on blood vessels and the heart through integrin receptors.
Nuclear Thyroid Hormone Receptors Thyroid hormones bind with high affinity to nuclear thyroid hormone receptors (TRs) α and β. Both TRα and TRβ are expressed in most tissues, but their relative expression levels vary among organs; TRα is particularly abundant in brain, kidneys, gonads, muscle, and heart, whereas TRβ expression is relatively high in the pituitary and liver. Both receptors are variably spliced to form unique isoforms. The TRβ2 isoform, which has a unique amino terminus, is selectively expressed in the hypothalamus and pituitary, where it plays a role in feedback control of the thyroid axis (see above). The TRα2 isoform contains a unique carboxy terminus that precludes thyroid hormone binding; it may function to block the action of other TR isoforms.
The TRs contain a central DNA-binding domain and a C-terminal ligand-binding domain. They bind to specific DNA sequences, termed thyroid response elements (TREs), in the promoter regions of target genes (Fig. 405-4). The receptors bind as homodimers or, more commonly, as heterodimers with retinoic acid × receptors (RXRs) (Chap. 400e). The activated receptor can either stimulate gene transcription (e.g., myosin heavy chain α) or inhibit transcription (e.g., TSH β-subunit gene), depending on the nature of the regulatory elements in the target gene.
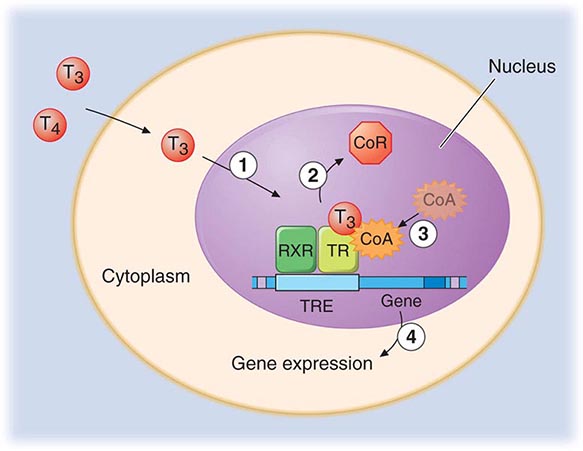
FIGURE 405-4 Mechanism of thyroid hormone receptor action. The thyroid hormone receptor (TR) and retinoid × receptor (RXR) form heterodimers that bind specifically to thyroid hormone response elements (TRE) in the promoter regions of target genes. In the absence of hormone, TR binds co-repressor (CoR) proteins that silence gene expression. The numbers refer to a series of ordered reactions that occur in response to thyroid hormone: (1) T4 or T3 enters the nucleus; (2) T3 binding dissociates CoR from TR; (3) co-activators (CoA) are recruited to the T3-bound receptor; and (4) gene expression is altered.
Thyroid hormones (T3 and T4) bind with similar affinities to TRα and TRβ. However, structural differences in the ligand binding domains provide the potential for developing receptor-selective agonists or antagonists, and these are under investigation. T3 is bound with 10–15 times greater affinity than T4, which explains its increased hormonal potency. Although T4 is produced in excess of T3, receptors are occupied mainly by T3, reflecting T4 → T3 conversion by peripheral tissues, greater T3 bioavailability in the plasma, and the greater affinity of receptors for T3. After binding to TRs, thyroid hormone induces conformational changes in the receptors that modify its interactions with accessory transcription factors. Importantly, in the absence of thyroid hormone binding, the aporeceptors bind to co-repressor proteins that inhibit gene transcription. Hormone binding dissociates the co-repressors and allows the recruitment of co-activators that enhance transcription. The discovery of TR interactions with co-repressors explains the fact that TR silences gene expression in the absence of hormone binding. Consequently, hormone deficiency has a profound effect on gene expression because it causes gene repression as well as loss of hormone-induced stimulation. This concept has been corroborated by the finding that targeted deletion of the TR genes in mice has a less pronounced phenotypic effect than hormone deficiency.
Thyroid Hormone Resistance Resistance to thyroid hormone (RTH) is an autosomal dominant disorder characterized by elevated thyroid hormone levels and inappropriately normal or elevated TSH. Individuals with RTH do not, in general, exhibit signs and symptoms that are typical of hypothyroidism because hormone resistance is partial and is compensated by increased levels of thyroid hormone. The clinical features of RTH can include goiter, attention deficit disorder, mild reduction in IQ, delayed skeletal maturation, tachycardia, and impaired metabolic responses to thyroid hormone.
Classical forms of RTH are caused by mutations in the TRβ gene. These mutations, located in restricted regions of the ligand-binding domain, cause loss of receptor function. However, because the mutant receptors retain the capacity to dimerize with RXRs, bind to DNA, and recruit co-repressor proteins, they function as antagonists of the remaining normal TRβ and TRα receptors. This property, referred to as “dominant negative” activity, explains the autosomal dominant mode of transmission. The diagnosis is suspected when unbound thyroid hormone levels are increased without suppression of TSH. Similar hormonal abnormalities are found in other affected family members, although the TRβ mutation arises de novo in about 20% of patients. DNA sequence analysis of the TRβ gene provides a definitive diagnosis. RTH must be distinguished from other causes of euthyroid hyperthyroxinemia (e.g., FDH) and inappropriate secretion of TSH by TSH-secreting pituitary adenomas (Chap. 403). In most patients, no treatment is indicated; the importance of making the diagnosis is to avoid inappropriate treatment of mistaken hyperthyroidism and to provide genetic counseling.
A distinct form of RTH is caused by mutations in the TRα gene. Affected patients have many clinical features of congenital hypothyroidism including growth retardation, skeletal dysplasia, and severe constipation. In contrast to RTH caused by mutations in TRβ, thyroid function tests include normal TSH, low or normal T4, and normal or elevated T3 levels. These distinct clinical and laboratory features underscore the different tissue distribution and functional roles of TRβ and TRα. Optimal treatment of patients with RTH caused by TRα mutations has not been established.
PHYSICAL EXAMINATION
In addition to the examination of the thyroid itself, the physical examination should include a search for signs of abnormal thyroid function and the extrathyroidal features of ophthalmopathy and dermopathy (see below). Examination of the neck begins by inspecting the seated patient from the front and side and noting any surgical scars, obvious masses, or distended veins. The thyroid can be palpated with both hands from behind or while facing the patient, using the thumbs to palpate each lobe. It is best to use a combination of these methods, especially when nodules are small. The patient’s neck should be slightly flexed to relax the neck muscles. After locating the cricoid cartilage, the isthmus, which is attached to the lower one-third of the thyroid lobes, can be identified and then followed laterally to locate either lobe (normally, the right lobe is slightly larger than the left). By asking the patient to swallow sips of water, thyroid consistency can be better appreciated as the gland moves beneath the examiner’s fingers.
Features to be noted include thyroid size, consistency, nodularity, and any tenderness or fixation. An estimate of thyroid size (normally 12–20 g) should be made, and a drawing is often the best way to record findings. However, ultrasound is the method of choice when it is important to determine thyroid size accurately. The size, location, and consistency of any nodules should also be defined. A bruit or thrill over the gland, located over the insertion of the superior and inferior thyroid arteries (supero- or inferolaterally), indicates increased vascularity, as occurs in hyperthyroidism. If the lower borders of the thyroid lobes are not clearly felt, a goiter may be retrosternal. Large retrosternal goiters can cause venous distention over the neck and difficulty breathing, especially when the arms are raised (Pemberton’s sign). With any central mass above the thyroid, the tongue should be extended, as thyroglossal cysts then move upward. The thyroid examination is not complete without assessment for lymphadenopathy in the supraclavicular and cervical regions of the neck.
LABORATORY EVALUATION
Measurement of Thyroid Hormones The enhanced sensitivity and specificity of TSH assays have greatly improved laboratory assessment of thyroid function. Because TSH levels change dynamically in response to alterations of T4 and T3, a logical approach to thyroid testing is to first determine whether TSH is suppressed, normal, or elevated. With rare exceptions (see below), a normal TSH level excludes a primary abnormality of thyroid function. This strategy depends on the use of immunochemiluminometric assays (ICMAs) for TSH that are sensitive enough to discriminate between the lower limit of the reference range and the suppressed values that occur with thyrotoxicosis. Extremely sensitive (fourth-generation) assays can detect TSH levels ≤0.004 mIU/L, but, for practical purposes, assays sensitive to ≤0.1 mIU/L are sufficient. The widespread availability of the TSH ICMA has rendered the TRH stimulation test obsolete, because the failure of TSH to rise after an intravenous bolus of 200–400 μg TRH has the same implications as a suppressed basal TSH measured by ICMA.
The finding of an abnormal TSH level must be followed by measurements of circulating thyroid hormone levels to confirm the diagnosis of hyperthyroidism (suppressed TSH) or hypothyroidism (elevated TSH). Radioimmunoassays are widely available for serum total T4 and total T3. T4 and T3 are highly protein-bound, and numerous factors (illness, medications, genetic factors) can influence protein binding. It is useful, therefore, to measure the free, or unbound, hormone levels, which correspond to the biologically available hormone pool. Two direct methods are used to measure unbound thyroid hormones: (1) unbound thyroid hormone competition with radiolabeled T4 (or an analogue) for binding to a solid-phase antibody, and (2) physical separation of the unbound hormone fraction by ultracentrifugation or equilibrium dialysis. Although early unbound hormone immunoassays suffered from artifacts, newer assays correlate well with the results of the more technically demanding and expensive physical separation methods. An indirect method that is now less commonly used to estimate unbound thyroid hormone levels is to calculate the free T3 or free T4 index from the total T4 or T3 concentration and the thyroid hormone binding ratio (THBR). The latter is derived from the T3-resin uptake test, which determines the distribution of radiolabeled T3 between an absorbent resin and the unoccupied thyroid hormone binding proteins in the sample. The binding of the labeled T3 to the resin is increased when there is reduced unoccupied protein binding sites (e.g., TBG deficiency) or increased total thyroid hormone in the sample; it is decreased under the opposite circumstances. The product of THBR and total T3 or T4 provides the free T3 or T4 index. In effect, the index corrects for anomalous total hormone values caused by abnormalities in hormone-protein binding.
Total thyroid hormone levels are elevated when TBG is increased due to estrogens (pregnancy, oral contraceptives, hormone therapy, tamoxifen, selective estrogen receptor modulators, inflammatory liver disease) and decreased when TBG binding is reduced (androgens, nephrotic syndrome). Genetic disorders and acute illness can also cause abnormalities in thyroid hormone binding proteins, and various drugs (phenytoin, carbamazepine, salicylates, and nonsteroidal anti-inflammatory drugs [NSAIDs]) can interfere with thyroid hormone binding. Because unbound thyroid hormone levels are normal and the patient is euthyroid in all of these circumstances, assays that measure unbound hormone are preferable to those for total thyroid hormones.
For most purposes, the unbound T4 level is sufficient to confirm thyrotoxicosis, but 2–5% of patients have only an elevated T3 level (T3 toxicosis). Thus, unbound T3 levels should be measured in patients with a suppressed TSH but normal unbound T4 levels.
There are several clinical conditions in which the use of TSH as a screening test may be misleading, particularly without simultaneous unbound T4 determinations. Any severe nonthyroidal illness can cause abnormal TSH levels (see below). Although hypothyroidism is the most common cause of an elevated TSH level, rare causes include a TSH-secreting pituitary tumor (Chap. 403), thyroid hormone resistance, and assay artifact. Conversely, a suppressed TSH level, particularly <0.01 mIU/L, usually indicates thyrotoxicosis. However, subnormal TSH levels between 0.01 and 0.1 mIU/L may be seen during the first trimester of pregnancy (due to hCG secretion), after treatment of hyperthyroidism (because TSH can remain suppressed for several months), and in response to certain medications (e.g., high doses of glucocorticoids or dopamine). Importantly, secondary hypothyroidism, caused by hypothalamic-pituitary disease, is associated with a variable (low to high-normal) TSH level, which is inappropriate for the low T4 level. Thus, TSH should not be used as an isolated laboratory test to assess thyroid function in patients with suspected or known pituitary disease.
Tests for the end-organ effects of thyroid hormone excess or depletion, such as estimation of basal metabolic rate, tendon reflex relaxation rates, or serum cholesterol, are not useful as clinical determinants of thyroid function.
Tests to Determine the Etiology of Thyroid Dysfunction Autoimmune thyroid disease is detected most easily by measuring circulating antibodies against TPO and Tg. Because antibodies to Tg alone are uncommon, it is reasonable to measure only TPO antibodies. About 5–15% of euthyroid women and up to 2% of euthyroid men have thyroid antibodies; such individuals are at increased risk of developing thyroid dysfunction. Almost all patients with autoimmune hypothyroidism, and up to 80% of those with Graves’ disease, have TPO antibodies, usually at high levels.
TSIs are antibodies that stimulate the TSH-R in Graves’ disease. They are most commonly measured by commercially available tracer displacement assays called TRAb (TSH receptor antibody) with the assumption that elevated levels in the setting of clinical hyperthyroidism reflect stimulatory effects on the TSH receptor. A bioassay is less commonly used. The main use of these assays is to predict neonatal thyrotoxicosis caused by high maternal levels of TRAb or TSI (>3× upper limit of normal) in the last trimester of pregnancy.
Serum Tg levels are increased in all types of thyrotoxicosis except thyrotoxicosis factitia caused by self-administration of thyroid hormone. Tg levels are particularly increased in thyroiditis, reflecting thyroid tissue destruction and release of Tg. The main role for Tg measurement, however, is in the follow-up of thyroid cancer patients. After total thyroidectomy and radioablation, Tg levels should be undetectable; in the absence of anti-Tg antibodies, measurable levels indicate incomplete ablation or recurrent cancer.
Radioiodine Uptake and Thyroid Scanning The thyroid gland selectively transports radioisotopes of iodine (123I, 125I, 131I) and 99mTc pertechnetate, allowing thyroid imaging and quantitation of radioactive tracer fractional uptake.
Nuclear imaging of Graves’ disease is characterized by an enlarged gland and increased tracer uptake that is distributed homogeneously. Toxic adenomas appear as focal areas of increased uptake, with suppressed tracer uptake in the remainder of the gland. In toxic MNG, the gland is enlarged—often with distorted architecture—and there are multiple areas of relatively increased (functioning nodules) or decreased tracer uptake (suppressed thyroid parenchyma or nonfunctioning nodules). Subacute, viral, and postpartum thyroiditis are associated with very low uptake because of follicular cell damage and TSH suppression. Thyrotoxicosis factitia is also associated with low uptake. In addition, if there is excessive circulating exogenous iodine (e.g., from dietary sources of iodinated contrast dye), the radionuclide uptake is low even in the presence of increased thyroid hormone production.
Thyroid scintigraphy is not used in the routine evaluation of patients with thyroid nodules, but should be performed if the serum TSH level is subnormal to determine if functioning thyroid nodules are present. Functioning or “hot” nodules are almost never malignant, and fine-needle aspiration (FNA) biopsy is not indicated. The vast majority of thyroid nodules do not produce thyroid hormone (“cold” nodules), and these are more likely to be malignant (~5–10%). Whole-body and thyroid scanning is also used in the treatment and surveillance of thyroid cancer. After thyroidectomy for thyroid cancer, the TSH level is raised by either using a thyroid hormone withdrawal protocol or recombinant human TSH injection (see below). Administration of 131I allows whole-body scanning (WBS) to confirm remnant ablation and to detect any functioning metastases. In addition, WBS may be helpful in surveillance of patients at risk for recurrence.
Thyroid Ultrasound Ultrasonography is valuable for the diagnosis and evaluation of patients with nodular thyroid disease (Table 405-4). Evidence-based guidelines recommend thyroid ultrasonography for all patients suspected of having thyroid nodules by either physical examination or another imaging study. Using 10- to 12-MHz linear transducers, resolution and image quality are excellent, allowing the characterization of nodules and cysts >3 mm. Certain sonographic patterns are highly suggestive of malignancy (e.g., hypoechoic solid nodules with infiltrative borders and microcalcifications), whereas other features correlate with benignity (e.g., spongiform nodules defined as those with multiple small internal cystic areas) (Fig. 405-5). In addition to evaluating thyroid nodules, ultrasound is useful for monitoring nodule size and for the aspiration of nodules or cystic lesions. Ultrasound-guided FNA biopsy of thyroid lesions lowers the rate of inadequate sampling and decreases sample error, thereby reducing the false-negative rate of FNA cytology. Ultrasonography of the central and lateral cervical lymph node compartments is indispensable in the evaluation thyroid cancer patients, preoperatively and during follow-up.
GRAYSCALE SONOGRAPHIC FEATURES ASSOCIATED WITH THYROID CANCER |
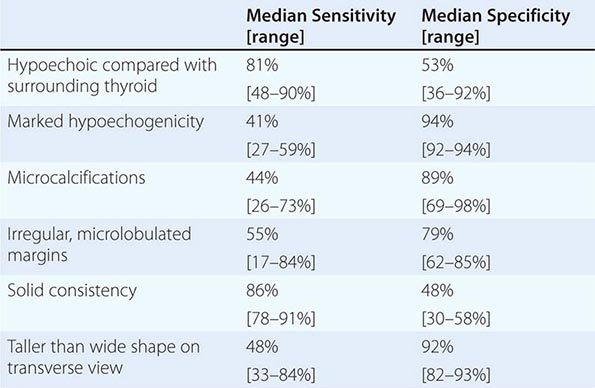
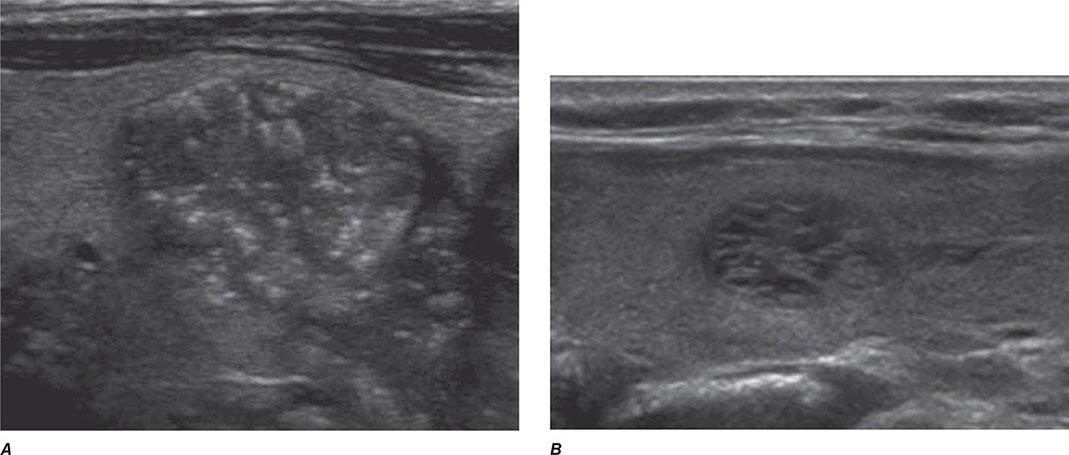
FIGURE 405-5 Sonographic patterns of thyroid nodules. A. High suspicion ultrasound pattern for thyroid malignancy (hypoechoic solid nodule with irregular borders and microcalcifications). B. Very low suspicion ultrasound pattern for thyroid malignancy (spongiform nodule with microcystic areas comprises over >50% of nodule volume).
HYPOTHYROIDISM
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


