Introduction
This chapter presents a general overview of the key hormones involved in the regulation of calcium, phosphate, and bone mineral metabolism. These include parathyroid hormone, vitamin D—principally the 1,25-(OH)2 vitamin D metabolite (1,25-dihydroxycholecalciferol)—calcitonin, and fibroblast growth factor (FGF)-23. The cycle of bone remodeling is described as a basis for understanding normal maintenance of skeletal integrity in adults and of mineral homeostasis. The symptoms and signs caused by excess or deficiency of the calciotropic hormones are presented along with the natural histories of primary hyperparathyroidism, familial (benign) hypocalciuric hypercalcemia, hypercalcemia of malignancy, different forms of hypoparathyroidism, and medullary carcinoma of the thyroid. Two of the most commonly encountered causes of low bone mass—osteoporosis and osteomalacia—are reviewed, along with discussions regarding their pathogenesis.
Normal Regulation of Calcium & Phosphorus Metabolism
Normal parathyroid glands each weigh 30–40 mg and are gray-tan to yellow-gray. Each individual typically has four glands, so that the average total parathyroid tissue mass in the adult is 120–160 mg.
The superior pair of parathyroid glands arise from the fourth branchial pouches in the embryo. These glands are located near the point of intersection of the middle thyroid artery and the recurrent laryngeal nerve. The superior parathyroid glands may be attached to the thyroid capsule posteriorly or, rarely, embedded in the thyroid gland itself. Alternative locations include the tracheoesophageal groove and the retroesophageal space. The blood supply to the superior parathyroid glands is from the inferior thyroid artery or, less commonly, the superior thyroid artery.
The inferior parathyroid glands develop from the third branchial pouch, as does the thymus gland. These glands typically lie at or near the lower pole of the thyroid gland lateral to the trachea. The inferior glands receive their blood supply from the inferior thyroid arteries. The location of the inferior parathyroid glands is variable. When there are ectopic glands, they are typically found in association with thymic remnants. A common site for ectopic glands is the anterior mediastinum. Less common ectopic locations are the carotid sheath, pericardium, and pharyngeal submucosa. About 10% of people have additional (supernumerary) parathyroid glands. This becomes a critically important issue when such ectopic glands develop hyperparathyroidism.
The parathyroid gland is composed of three different cell types: chief cells, clear cells, and oxyphil cells. Chief cells are small in diameter (4–8 μm) with central nuclei and are thought to be responsible for the synthesis and secretion of parathyroid hormone (PTH). In their active state, they have a prominent endoplasmic reticulum and dense Golgi regions where PTH is synthesized and packaged for secretion. Clear cells are probably chief cells with an increased glycogen content. Oxyphil cells appear in the parathyroid glands after puberty. They are larger than chief cells (6–10 μm), and their number increases with age. It is not clear whether these cells secrete PTH and whether they are derived from chief cells.
The normal adult parathyroid gland contains fat. The relative contribution of fat to the glandular mass increases with age and may reach 60–70% of gland volume in the elderly. If hyperplasia or adenomatous changes occur, the glandular fat content decreases dramatically.
Approximately 99% of total body calcium is found in the skeleton and teeth; the remainder is in the extracellular fluids. Calcium in these fluids exists in three forms: ionized, protein bound, and complexed. About 47% of total blood calcium is protein bound, predominantly to albumin but also to globulins. A similar fraction is ionized. The remainder is complexed to organic ions such as citrate, phosphate, and bicarbonate. Serum ionized calcium controls vital cellular functions such as hormone secretion and action, muscle contraction, neuromuscular transmission, and blood clotting. The binding of calcium to albumin is pH dependent, increasing with alkalosis and decreasing with acidosis. Thus, if the ionized calcium is low, acidosis tends to protect against symptomatic hypocalcemia. Conversely, alkalosis predisposes to symptomatic hypocalcemia.
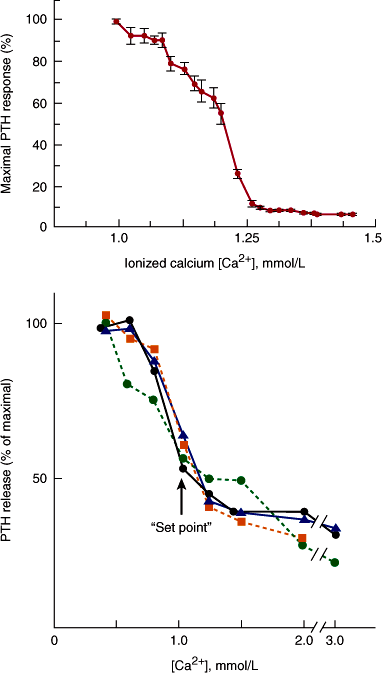
Circulating levels of PTH can change within seconds after an alteration in serum calcium. PTH secretory rates are related to the serum ionized calcium concentration by an inverse sigmoidal relationship (Figure 17–1). Low ionized calcium concentrations maximally stimulate secretion, whereas increases in calcium suppress the production and release of PTH. PTH secretion is exquisitely sensitive to very small changes in the calcium concentration, which have substantial effects on the rate of hormone synthesis and release.
Figure 17–1
Inverse sigmoidal relationship between parathyroid hormone (PTH) release and the extracellular calcium concentration in human studies (upper panel) and in vitro in human parathyroid cells (bottom panel). Studies shown in the upper panel were performed by infusing calcium and the calcium chelator EDTA into normal subjects. Serum intact PTH was measured by a two-site immunoradiometric assay. In the lower panel, PTH was measured in the medium surrounding parathyroid cells in vitro by an assay for intact PTH. The midpoint between the maximal and minimal secretory rates is defined as the set point for secretion. (Redrawn, with permission, from Brown E. Extracellular Ca2+ sensing, regulation of parathyroid cell function, and role of Ca2+ and other ions as extracellular [first] messengers. Physiol Rev. 1991;71:371.)
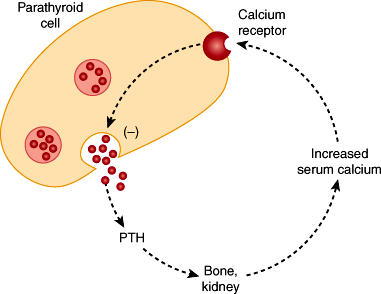
The extracellular calcium-sensing receptor (CaSR) is expressed by parathyroid and many other types of cells. Its job is to detect changes in the extracellular calcium concentration. This receptor is activated by increases in the calcium concentration and couples to intracellular pathways, which inhibits hormone secretion (Figure 17–2) and parathyroid cell proliferation. CaSRs are also expressed in the kidney, thyroid C cells, brain, and many other tissues. Hypocalcemia is also sensed by the CaSR, and PTH secretion is stimulated. Chronic hypocalcemia stimulates proliferation of parathyroid cells, which eventually results in glandular hyperplasia. Thus, the CaSR controls secretion and proliferation in appropriate directions to respond to physiologic needs.
Figure 17–2
Sequence of events by which the calcium ion concentration is sensed by the parathyroid calcium-sensing receptor (CaSR). Activation of this receptor is eventually linked through intracellular signal transduction pathways to the inhibition of PTH secretion and parathyroid cell proliferation. (Redrawn with modification from Taylor R. A new receptor for calcium ions. J NIH Res. 1994;6:25.)
PTH is produced in the parathyroid glands as a 115-amino-acid precursor molecule (preproPTH) that is successively cleaved within the cell to form the mature 84-amino-acid peptide PTH(1–84) (Figure 17–3). This form of the hormone is packaged into secretory granules and released into the circulation. PTH(1–84) is the biologically active form of PTH at target cells and has a very short half-life in vivo of approximately 10 minutes. PTH(1–84) is metabolized in the liver and other tissues to midregion and carboxyl terminal forms that are probably biologically inactive. These circulating fragments accumulate to very high levels in patients with renal failure, because the kidney is an important site for clearance of PTH from the body. Intact PTH assays in routine use measure PTH(1–84) using immunoradiometric or immunochemiluminometric methods that employ two antibodies: one directed against an amino terminal epitope, which is labeled, and the other directed against a carboxyl terminal epitope of PTH(1–84), which is immobilized (Figure 17–4). It is now clear that these “intact” PTH assays also detect amino-terminally truncated fragments of hormone such as PTH(7–84) that accumulate particularly in the serum of uremic patients. It is estimated that 30–50% of circulating “intact PTH” in uremic sera may represent these amino terminal fragments. This led to the development of “whole PTH” assays that only detect PTH(1–84). The amino terminal antibody in these assays specifically recognizes the first six amino acids of PTH(1–84). Such assays, however, have not replaced the original intact assays for routine clinical use.
Figure 17–3
Biosynthetic events in the production of parathyroid hormone (PTH) within the parathyroid cell. PreproPTH gene is transcribed to its mRNA, which is translated on the ribosomes to preproPTH (amino acids −29 to +84). The presequence is removed within the endoplasmic reticulum, yielding proPTH (−6 to +84). An additional six-amino-acid fragment is removed in the Golgi. Mature PTH(1−84) released from the Golgi is packaged in secretory granules and released into the circulation in the presence of hypocalcemia. The calcium-sensing receptor (CaSR) or CaR is proposed to sense changes in extracellular calcium that affect both the release of PTH and the transcription of the preproPTH gene. High extracellular calcium concentrations also promote the intracellular degradation of PTH. (Redrawn, with permission, from Habener JF et al. Biosynthesis of parathyroid hormone. Recent Prog Horm Res. 1977;33:249.)
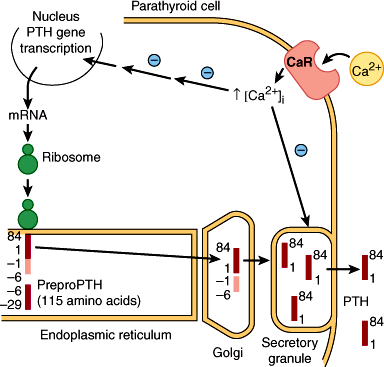
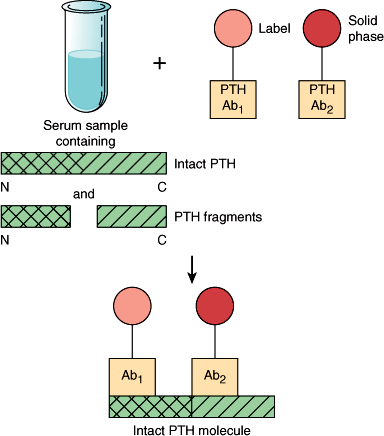
Figure 17–4
Schematic representation of the principle of the two-site assay for parathyroid hormone (PTH), in this case full-length, biointact PTH(1–84). The label may be a luminescent probe or 125I in the immunochemiluminometric or immunoradiometric assay, respectively. Two different region-specific antibodies are used (Ab1 and Ab2). The epitope for Ab1 is at the extreme N-terminus ensuring that only the hormone species containing both N- and C-terminal/midregion immunodeterminants are counted in the assay.
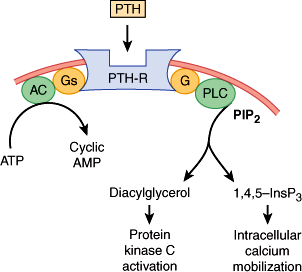
There are two types of PTH receptors. The type 1 receptor recognizes PTH and parathyroid hormone–related peptide (PTHrP) and is also called the PTH-1 receptor. The type 2 receptor is specific for PTH. PTH and PTHrP (described later) bind to the type 1 receptor through residues in their amino terminal domains. PTH activates adenylyl cyclase and produces the second-messenger cAMP (Figure 17–5). The type 1 receptor also couples to the stimulation of phospholipase C activity, leading to the generation of inositol trisphosphate and diacylglycerol (Figure 17–5). Activation of this signal transduction pathway induces intracellular calcium mobilization and protein kinase C activation in PTH- and PTHrP-responsive cells. The type 2 PTH receptor is expressed in nonclassic PTH target tissues (ie, brain, pancreas, testis, and placenta). This receptor is not thought to be involved in mineral balance, and its natural ligand may be a hypothalamic peptide called tubuloinfundibular peptide.
Figure 17–5
Signal transduction pathways activated by parathyroid hormone (PTH) binding to the PTH-1 receptor (PTH-R) in a target cell. PTH interacts with its receptor. This enhances guanosine triphosphate binding to the stimulatory G protein of adenylyl cyclase Gs, which activates the enzyme. Cyclic adenosine monophosphate (cAMP) is formed. PTH also increases G protein-dependent activation of phospholipase C (PLC), which catalyzes the breakdown of the membrane phospholipid phosphatidylinositol 4,5-bisphosphate (PIP2). This produces the second messengers, inositol trisphosphate (1,4,5-InsP3) and diacylglycerol. 1,4,5-InsP3 mobilizes intracellular calcium, and diacylglycerol activates protein kinase C.
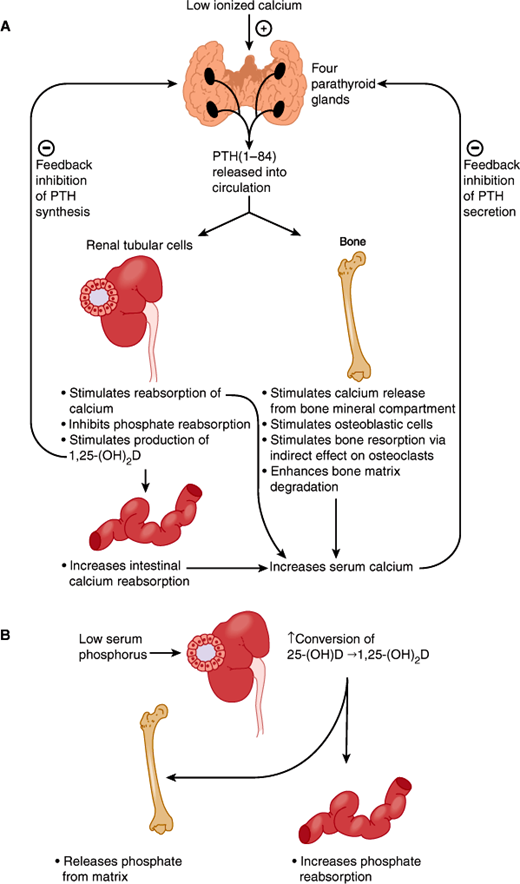
The serum ionized calcium and phosphate concentrations reflect the net transfer of these ions from bone, GI tract, and glomerular filtrate. PTH and 1,25-(OH)2D play key roles in the regulation of calcium and phosphate balance (Figure 17–6). When the serum calcium concentration falls, PTH is rapidly released and acts quickly to promote calcium reabsorption in the distal tubule and the medullary thick ascending limb of Henle loop. PTH also stimulates the release of calcium from bone. These actions serve to restore serum calcium levels to normal.
Figure 17–6
Main actions of parathyroid hormone (PTH) and 1,25-(OH)2D in the maintenance of calcium and phosphate homeostasis. (Redrawn, with permission, from Chandrasoma P et al, eds. Concise Pathology, 3rd ed. Originally published by Appleton & Lange. Copyright © 1998 by The McGraw-Hill Companies, Inc.)
The renal action of PTH is rapid, occurring within minutes after an increase in the hormone. The overall effect of PTH on the kidney, however, depends on several factors. When hypocalcemia is present and PTH is elevated, urinary calcium excretion is low. This reflects the full expression of the primary renal effect of PTH to enhance renal calcium reabsorption. When PTH levels are high in primary hyperparathyroidism, hypercalcemia results from increased mobilization of calcium from bone and enhanced intestinal calcium absorption. These events increase the delivery of calcium to the glomerular filtrate. Because more calcium is filtered, more is excreted in the urine, despite the high PTH levels. If the filtered load of calcium is normal or low in a patient with primary hyperparathyroidism—because of a low dietary calcium intake or demineralized bone—urinary calcium excretion may be normal or even low. Thus, there may be considerable variability in calcium excretion among patients with hyperparathyroidism.
If kidney function is normal, chronic elevation in serum PTH increases renal 1,25-(OH)2D production. This steroid hormone stimulates both calcium and phosphate absorption across the small intestine (Figure 17–6). The effect requires at least 24 h to develop fully and begin to restore normal calcium levels. Achievement of eucalcemia then leads to a downward readjustment in the PTH secretory rate. Any increase in 1,25-(OH)2D serves to inhibit further PTH synthesis by binding to vitamin D receptors in the parathyroid.
The major effect of PTH on phosphate handling is to promote its excretion by inhibition of sodium-dependent phosphate transport in the proximal tubule. Serum phosphate levels are thought to affect PTH secretion rates directly, with hyperphosphatemia serving as a stimulus to PTH secretion by an uncertain mechanism. Hypophosphatemia enhances the conversion of 25-(OH)D to 1,25-(OH)2D in the kidney, which through its intestinal and renal effects promotes phosphate retention. Hyperphosphatemia also inhibits 1,25-(OH)2D production (see below) and lowers serum calcium by complexing with it in the circulation.
PTH also increases urinary excretion of bicarbonate through its action on the proximal tubule. This can produce proximal renal tubular acidosis. These physiologic responses to PTH are the basis for the hypophosphatemia and hyperchloremic acidosis commonly observed in patients with hyperparathyroidism. Dehydration is also common in moderate to severe hypercalcemia of any origin. This is due to the effect of hypercalcemia on vasopressin action in the medullary thick ascending limb of the kidney. High calcium levels, presumably by interacting with renal CaSRs, blunt the ability of endogenous vasopressin to stimulate water reabsorption. Thus, hypercalcemia induces vasopressin-resistant nephrogenic diabetes insipidus.
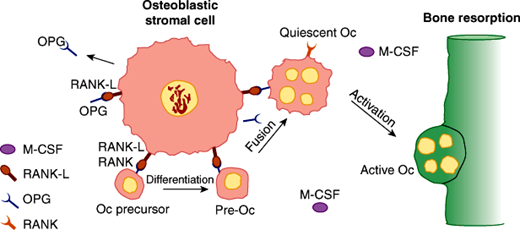
In conjunction with 1,25-(OH)2D, PTH increases bone resorption to restore normocalcemia (see below). PTH enhances osteoclastic activity through the stimulation of RANK-L (receptor activator of nuclear factor kappa B ligand), which is expressed by cells of the osteoblastic lineage (including stromal cells and osteoblasts). RANK-L interacts with its receptor RANK on cells of the osteoclast lineage to stimulate their differentiation and function, which is bone resorption (Figure 17–7). Once resorption ceases, bone formation ensues, because the processes of resorption and formation are coupled. In primary and secondary hyperparathyroidism, when PTH production rates are excessive, net bone loss may occur over time, perhaps because even though the processes of formation and resorption are coupled, they may not occur with 100% efficiency.
Figure 17–7
Cell-cell interactions and molecules essential for the differentiation and activation of osteoclasts. A cell surface molecule known as RANK-L on osteoblastic bone marrow stromal cells can interact with osteoclastic precursor cells in the bone marrow (derived from cells of the monocytic lineage) through their cell surface molecules designated RANK. This interaction, in the presence of sufficient macrophage colony-stimulating factor (mCSF), promotes the differentiation and fusion of these cells eventually to form mature osteoclasts and enables otherwise quiescent osteoclasts to resorb bone. These pathways are interfered with by the elaboration of a secreted decoy receptor molecule for RANK-L known as OPG, which blocks activation and differentiation of osteoclasts. (Oc, osteoclast.) (Redrawn, with permission, from Goltzman D. Osteolysis and cancer. J Clin Invest. 2001;107:1219.)
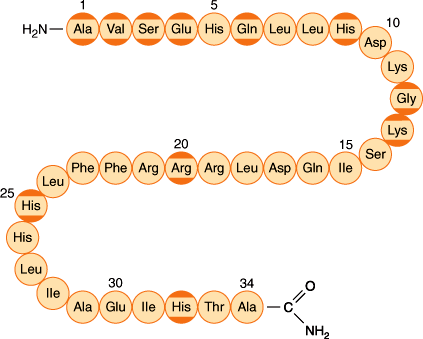
PTHrP is a 141-amino-acid peptide that is homologous with PTH at its amino terminal region (Figure 17–8) and is recognized by the type 1 PTH receptor. Consequently, PTHrP has effects on bone and kidney similar to those of PTH; it increases bone resorption, increases phosphate excretion, and decreases renal calcium excretion. PTHrP is secreted by tumor cells and was originally identified as the cause of hypercalcemia of malignancy, a syndrome that can mimic primary hyperparathyroidism (see later).
Figure 17–8
The amino acid sequence of the 34 amino acid residue at the N terminal parathyroid hormone (PTH)–related peptide. Amino acids that are identical to those in PTH are shown with dark yellow borders. (Redrawn, with permission, from Felig P et al, eds. Endocrinology and Metabolism, 3rd ed. McGraw-Hill, 1995.)
Unlike PTH, which is exclusively produced by parathyroid cells, PTHrP is produced in many tissues. It functions mainly as a tissue growth and differentiation factor at the local level and a regulator of smooth muscle tone. In the normal development of cartilage and bone, PTHrP stimulates the proliferation of chondrocytes and inhibits the mineralization of cartilage. Embryos without PTHrP are nonviable, with multiple abnormalities of bone and cartilage. PTHrP also appears to regulate the normal development of skin, hair follicles, teeth, and the breast. PTHrP plays an important role in determining the calcium content of the milk from lactating animals.
Despite PTHrP binding to the same PTH-1 receptor (see above) to achieve most of its physiologic effects, new studies indicate that the consequences of PTH and PTHrP interacting with the receptor are surprisingly different. Each peptide has different effects on the conformational state and the extent of activation of the receptor. PTHrP can also be transcribed from a promoter that bypasses the signal peptide. This allows PTHrP (and not PTH) to enter the nucleus and mediate additional biological effects there. Thus, there are several ways by which cells can differentially react to these similar peptides.
Checkpoint
Describe the cell types in the parathyroid gland.
How do serum albumin concentration and blood pH influence the distribution of calcium into ionized and protein-bound fractions?
What advances have occurred in two-site immunoassays for PTH that affect uremic patients?
What are the actions of PTH and 1,25-(OH)2D on bone, kidney, and the GI tract?
What is PTHrP? How is its action similar to and different from that of PTH?
Bone has two compartments. On the outside is cortical or compact bone, which makes up 80% of the skeletal mass and plays a significant role in giving bone its strength. The other compartment is trabecular or cancellous bone, which makes up 20% of skeletal mass. Trabecular bone consists of interconnected plates, the trabeculae, which are covered by bone cells and are sites of active remodeling. The spaces in this irregular honeycomb are filled with bone marrow: either red marrow, in which hematopoiesis is active, or white marrow, which is mainly fat. Because of its high surface-to-volume ratio and abundant cellular activity, trabecular bone is remodeled more rapidly than cortical bone. Because of the low ratio of surface to volume, cortical bone is remodeled slowly.
To understand the remodeling process, it is important to know something about bone cells.
Osteocytes, the most abundant cells in bone, are derived from the osteoblast lineage and reside deep in the matrix. Osteocytes function as mechanoreceptors, detecting strain on the bone and signaling changes in bone remodeling. Osteoclasts, multinucleated giant cells specialized for resorption of bone, are terminally differentiated cells that arise continuously from hematopoietic precursors in the macrophage/monocyte lineage. The formation of osteoclasts requires the hematopoietic growth factor macrophage colony-stimulating factor (m-CSF) and a signal from marrow stromal cells. The critical signal, RANK-L, either resides on the surface of bone marrow stromal cells and osteoblastic cells or is secreted in the extracellular environment. This molecule, which is required for osteoclast differentiation and activation, binds to its receptor RANK on osteoclast precursors and signals to the cell interior. A variety of cells, including those from the marrow, produce a soluble, secreted decoy receptor, osteoprotegerin (OPG), that binds RANK-L, thereby preventing its interaction with RANK and halting osteoclast differentiation and activation (Figure 17–7). As osteoclasts mature, they acquire the capacity to produce osteoclast-specific enzymes and fuse to produce the mature multinucleated cell. The maturation process is accelerated by bone-resorbing hormones such as PTH and 1,25-(OH)2D, presumably through their effects on the RANK-L/OPG system.
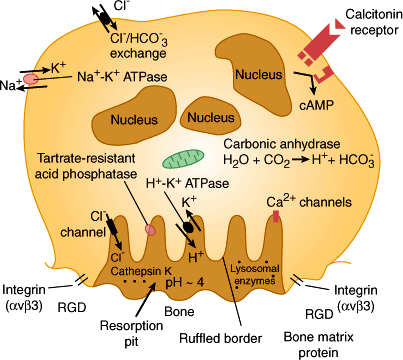
To resorb bone, the motile osteoclast alights on a bone surface and seals off an area by forming an adhesive ring in which cellular integrins bind tightly to bone matrix proteins (Figure 17–9). Having isolated an area of bone surface, the osteoclast develops above the surface an elaborately invaginated plasma membrane structure called the ruffled border. The ruffled border is a distinctive organelle, but it acts essentially as a huge lysosome, which dissolves bone mineral by secreting acid onto the isolated bone surface, and simultaneously breaks down the bone matrix by secretion of collagenase and proteases. One important protease is cathepsin K, an enzyme being studied as a potential target for the pharmacologic treatment of bone loss. The resulting collagen peptides have pyridinoline cross-links that can be assayed in urine as a measure of bone resorption rates. Bone resorption can be controlled in two ways: by regulating the formation of osteoclasts and by regulating the activity of mature osteoclasts. The osteoblast, or bone-forming cell, arises from a mesenchymal precursor induced to differentiate in the bone marrow stroma. When actively forming bone, the osteoblast is a tall, plump cell with an abundant Golgi apparatus. On active bone-forming surfaces, osteoblasts are found side by side, laying down bone matrix by secreting proteins and proteoglycans. The most important protein of bone matrix is type I collagen, which makes up 90% of bone matrix and is deposited in regular layers that serve as the main scaffold for deposition of minerals.
Figure 17–9
Schematic view of an active osteoclast. Calcitonin receptors, the ruffled border, and enzymes and channels involved in secretion of acid onto the bone surface are shown. Integrins (alpha V, beta 3) are transmembrane-spanning receptors on osteoclasts, which bind to determinants (RGD) in bone matrix proteins such as fibronectins. The integrins are responsible for the tight attachment of osteoclasts to the bone surface. Cathepsin K and other lysosomal enzymes are secreted into the resorption pit to dissolve the matrix. (Redrawn, with permission, from Felig P et al, eds. Endocrinology and Metabolism, 3rd ed. McGraw-Hill, 1995.)
After laying down bone matrix, osteoblasts mineralize it by depositing hydroxyapatite crystals in an orderly array on the collagen layers to produce lamellar bone. The process of mineralization is poorly understood but requires an adequate supply of extracellular calcium and phosphate as well as the enzyme alkaline phosphatase, which is secreted in large amounts by active osteoblasts.
Bone remodeling occurs in an orderly cycle in which old bone is resorbed and new bone is deposited. Cortical bone is remodeled from within by cutting cones (Figure 17–10), groups of osteoclasts that cut tunnels through the compact bone. They are followed by trailing osteoblasts, lining the tunnels and laying down a cylinder of new bone on their walls, so that the tunnels are progressively narrowed until all that remains are the tiny haversian canals, by which the cells that are left behind as resident osteocytes are fed.
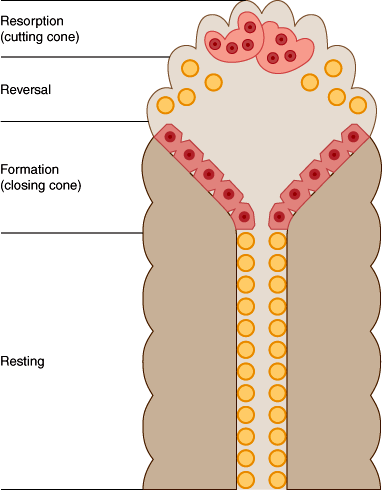
In trabecular bone, the remodeling process occurs on the surface (Figure 17–11). Osteoclasts first excavate a pit, and the pit is then filled in with new bone by osteoblasts. In a normal adult, this cycle takes approximately 200 days. At each remodeling site, bone resorption and new bone formation are ordinarily tightly coupled, so that in a state of zero net bone balance, the amount of new bone formed is precisely equivalent to the amount of old bone resorbed. This degree of balance is brief, however. From approximately 20 to 30 years of age, bone mass is consolidated after gains in growth and mineral deposition that were achieved during adolescence. After age 30 or 35 years, adult females begin to lose bone slowly.
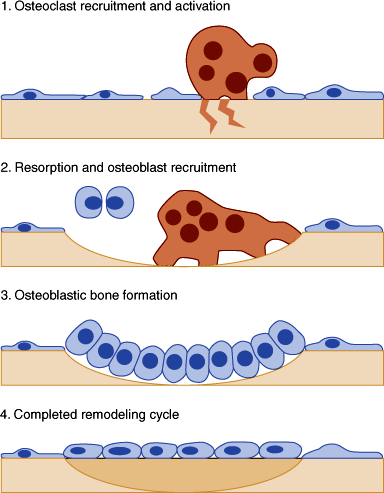
How osteoclasts and osteoblasts communicate to achieve the coupling that ensures perfect (or near-perfect) bone balance is not fully known. It appears that the important signals are local, not systemic. Although they have not been identified with certainty, one candidate is RANK-L (described above). RANK-L on the cell surface or as a soluble molecule binds to osteoclast precursors and supports their development and differentiation. RANK-L also binds to RANK on mature osteoclasts, and this may mediate the coupling of bone formation and bone resorption. The process of bone remodeling does not absolutely require systemic hormones except to ensure an adequate supply of calcium and phosphate. However, systemic hormones use the bone as a source of minerals for regulation of extracellular calcium homeostasis. Osteoblasts have receptors for PTH and 1,25-(OH)2D, but osteoclasts do not. Isolated osteoclasts do not respond to PTH or vitamin D, except in the presence of osteoblasts. This coupling mechanism makes certain that when bone resorption is activated by PTH (eg, to provide calcium to correct hypocalcemia) bone formation will also increase, tending to replenish lost bone.
Checkpoint
Describe the two compartments of bone.
How is bone resorption by osteoclasts controlled?
What is the role of osteoblasts in bone formation? How are the actions of osteoblasts and osteoclasts coupled?
Vitamin D is actually a prohormone produced in the dermis in response to ultraviolet B (UVB) exposure and metabolized to its active forms in the liver first, then in the kidney. The amount of sunlight exposure necessary to produce sufficient vitamin D is difficult to estimate because of individual differences in skin pigmentation, latitude, and time of day. Dietary sources are relatively modest in vitamin D content. For example, fish ingest ultraviolet-irradiated sterols (in phytoplankton and zooplankton) that are converted to vitamin D and stored in their livers.
7-Dehydrocholesterol, stored in the epidermis, is converted to vitamin D3 (cholecalciferol) by ultraviolet light (wavelengths 280–310 nm) (Figure 17–12). This step involves breakage of the B ring of the cholesterol structure to produce a secosteroid; hormones with an intact cholesterol nucleus (eg, estrogen) are called steroids. A similar process occurs in plants with one small structural difference, resulting in vitamin D2 rather than vitamin D3. Vitamin D2 is activated similarly to D3 in humans but does appear to have a decreased binding affinity for vitamin D–binding protein, resulting in enhanced clearance. This is particularly evident when large intermittent doses (ie, once weekly) rather than single daily doses are used medically in the treatment of vitamin D deficiency.
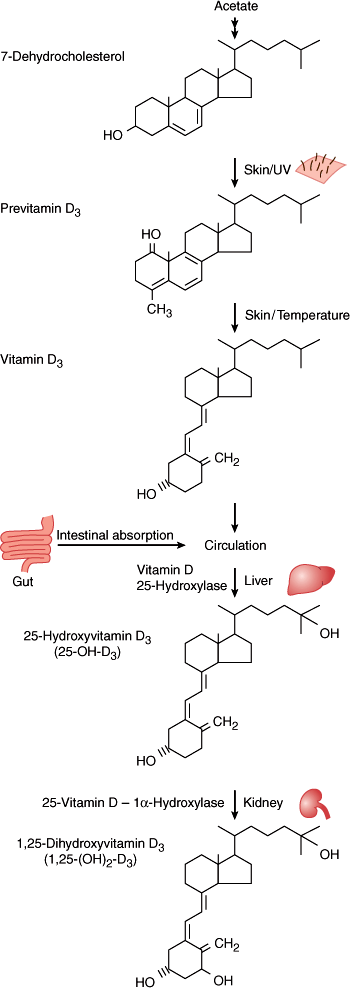
Although cutaneous synthesis of vitamin D can be sufficient to prevent rickets (the overt skeletal manifestation of vitamin D deficiency), it is not clear that sunlight exposure can be obtained in sufficient quantities to optimize vitamin D stores without untoward skin consequences. Further, at most latitudes in the United States, there is insufficient UVB radiation in the sunlight during the winter months to induce cutaneous production of vitamin D. In 2011, the Institute of Medicine revised the recommended intakes of vitamin D, recommending consumption of 400 IU/d up to 1 year of age, 600 IU/d for individuals 1–70 years of age, and 800 IU/d for individuals older than 70 years. In the United States, milk is supplemented with 400 IU of vitamin D per quart. Dietary supplements of vitamin D consist of vitamin D2 (ergocalciferol) and vitamin D3 (cholecalciferol).
While there is minimal regulation of vitamin D production in the skin, enhanced sun exposure does not result in vitamin D toxicity, as there is photo-conversion of vitamin D to inactive metabolites when skin levels of vitamin D rise. Vitamin D formed in the skin is a lipophilic substance that is transported to the liver bound to albumin and a specific vitamin D–binding protein (DBP). Ingested vitamin D is transported to the liver via chylomicrons. In the liver, vitamin D is hydroxylated to produce 25-hydroxyvitamin D (25-[OH]D) (Figure 17–12). This process is not closely regulated. 25-(OH)D is transported by DBP in the serum to target tissues and is stored in the liver and adipose tissues. The clinical test for vitamin D deficiency is measurement of the serum level of 25-(OH)D.
The final metabolic processing step in the synthesis of the circulating active hormone, 1,25-(OH)2D, takes place principally in the kidney, although many tissues can locally activate vitamin D for paracrine and autocrine functions. The conversion of 25-(OH)D to 1,25-(OH)2D by the 25-(OH)D 1-hydroxylase in the renal cortex is tightly regulated. The synthesis of 1,25-(OH)2D is increased by PTH, thus linking the formation of 1,25-(OH)2D closely to PTH in the integrated control of calcium homeostasis. The production of 1,25-(OH)2D is also stimulated by hypophosphatemia and hypocalcemia. On the other hand, hypercalcemia, hyperphosphatemia, fibroblast growth factor (FGF)-23, and decreased PTH will reduce 1,25-(OH)2D production. As an additional control, 1,25-(OH)2D induces the enzyme 24-hydroxylase, which catabolizes 25-(OH)D and 1,25-(OH)2D, thus reducing their levels. The coordinated control by PTH, blood mineral levels, and the vitamin D supply is very efficient. Serum levels of 1,25-(OH)2D vary only slightly over an enormous range of vitamin D production rates but respond precisely to changes in the serum levels of calcium and phosphate within the normal range.
The vitamin D receptor is a member of the steroid receptor superfamily of nuclear DNA-binding receptors. Upon ligand binding, the receptor attaches to enhancer sites in target genes and directly regulates their transcription. Thus, many of the effects of vitamin D involve new RNA and protein synthesis. Although many vitamin D metabolites are recognized by the receptor, 1,25-(OH)2D has an affinity approximately 1000-fold greater than that of 25-(OH)D. 25-(OH)D is present in the circulation at nanogram quantities, whereas 1,25-(OH)2D circulates in picogram quantities; thus, other vitamin D metabolites besides 1,25-(OH)2D may interact with the vitamin D receptor to produce clinical effects.
The primary target organs for 1,25-(OH)2D are intestine and bone. The most essential action of 1,25-(OH)2D is to stimulate the active intestinal transport of calcium in the duodenum. Calcium also can be absorbed passively through a paracellular route throughout the small intestine. However, particularly at low calcium intakes, the majority of gastrointestinal calcium absorption is mediated by the active vitamin D–mediated process. 1,25-(OH)2D also induces the active transport of phosphate, but passive absorption dominates this process, and the net effect of 1,25-(OH)2D is small.
In bone, 1,25-(OH)2D regulates a number of osteoblastic functions. Vitamin D deficiency leads to rickets, a defect in mineralization. However, the defect in mineralization results mainly from decreased delivery of calcium and phosphate to sites of mineralization. 1,25-(OH)2D also stimulates osteoclasts to resorb bone, releasing calcium to maintain the extracellular calcium concentration. This likely results from activation of the RANK-L/RANK signaling pathway by 1,25-(OH)2D.
To demonstrate the interplay among calcium, phosphorus, PTH, and vitamin D, consider a person who switches from a high normal to a low intake of calcium and phosphate: from 1200 to 300 mg/day of calcium (the equivalent of leaving three glasses of milk out of the diet). The net absorption of calcium falls sharply, causing a transient decrease in the serum calcium level. This activates a homeostatic response led by an increase in PTH. The increased PTH level stimulates the release of calcium from bone and the retention of calcium by the kidney. In addition, the increase in PTH, the fall in calcium, and the concomitant fall in the serum phosphate level (because of both decreased intake and PTH-induced phosphaturia) activate renal 1,25-(OH)2D synthesis. 1,25-(OH)2D increases the fraction of calcium that is absorbed from the intestine, further increases calcium release from bone, and restores the serum calcium to normal. 1,25-(OH)2D also promotes the intestinal absorption of phosphorus, although phosphorus absorption is much less regulated than calcium absorption. While these mechanisms can compensate for a low dietary calcium intake and maintain normal serum calcium and phosphorus levels, this is at the expense of mobilizing stored calcium from bone and maintaining an elevated PTH level. Over the long term, these compensatory mechanisms will result in depletion of skeletal calcium, increased bone resorption, and compromised skeletal integrity.
FGF-23 is a member of the large family of FGFs, local factors that are important in the control of cell proliferation and differentiation. FGF-23, in contrast to other FGF family members, plays a central role in the regulation of systemic phosphate homeostasis, vitamin D metabolism, and bone mineralization. Studies of kindreds with rare genetic disorders as well as transgenic and knockout mouse models that target essential molecules in FGF-23 signaling cascades have demonstrated the importance of FGF-23 in phosphate metabolism and skeletal mineralization.
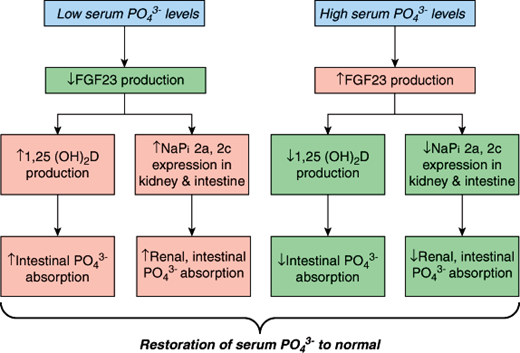
FGF-23 is produced by many tissues in the body, but its primary source appears to be bone cells, particularly osteocytes. A critical regulator of FGF-23 production is the serum phosphate level (Figure 17–13). Under normal physiologic conditions, when phosphate levels rise (eg, high-phosphate diet, renal failure), FGF-23 levels increase. When serum phosphate levels fall (eg, phosphate depletion, low-phosphate diet), serum FGF-23 levels decrease. In states of phosphate excess, FGF-23 reduces the expression of the sodium phosphate co-transporters (NaPi 2a and 2c) in the kidney and intestine. This leads to the rapid excretion of phosphate by the kidney and reduced intestinal phosphate absorption, which in turn restore the serum phosphate level to normal. To further control the amount of phosphate being delivered to the circulation, FGF-23 also inhibits the renal production of 1,25-(OH)2D (see Figure 17–13), further decreasing intestinal phosphorus absorption. These direct actions of FGF-23 are mediated by FGF receptors and their co-receptor transmembrane protein klotho.
Figure 17–13
Phosphate homeostasis is maintained by the coordinated actions of FGF-23 and 1,25(OH)2D. Low serum phosphate (PO43−) levels suppress FGF-23 production, which increases 1,25(OH)2D production and the expression of renal and intestinal phosphate transporters (NaPi 2a, 2c). As a result, intestinal and renal phosphate reabsorption rise to restore serum phosphate back to normal. When serum phosphate levels increase, FGF-23 levels rise, thereby suppressing these same biochemical pathways, and restoring serum phosphate balance.
Several rare disorders have served to define the actions of FGF-23 in phosphate and vitamin D metabolism in humans. Disorders of FGF-23 excess include X-linked hypophosphatemic rickets, autosomal dominant hypophosphatemic rickets, and tumor-induced osteomalacia (Table 17–12 and see the section on osteomalacia, below). Hypophosphatemia and osteomalacia resulting from phosphate wasting with a low or inappropriately normal serum 1,25-(OH)2D level are the hallmarks of these disorders. In contrast, loss of function of FGF-23, due to rare genetic disorders, is associated with syndromes of ectopic calcification, abnormal mineralization, and hyperphosphatemia. The role of FGF-23 in the hyperphosphatemia and osteodystrophy of chronic kidney disease is being actively investigated.
Checkpoint
How is vitamin D produced from 7-dehydrocholesterol?
Where is vitamin D stored?
Where does the final step in the activation of vitamin D take place, and how is it regulated?
What are the actions of vitamin D?
C cells of the thyroid gland secrete the peptide hormone calcitonin. They constitute 0.1% or less of thyroid cell mass and are distributed in the central parts of the lateral lobes of the thyroid, especially between the upper and middle thirds of the lobes. C cells are neuroendocrine cells derived from the ultimobranchial body, a structure that fuses with the thyroid.
C cells are small spindle-shaped or polygonal cells distributed throughout the thyroid. They contain abundant granules, mitochondria, and Golgi. They may be present as single cells or arranged in nests, cords, and sheets within the thyroid parenchyma. They are often found within thyroid follicles, are larger than follicular cells, and stain positively for calcitonin.
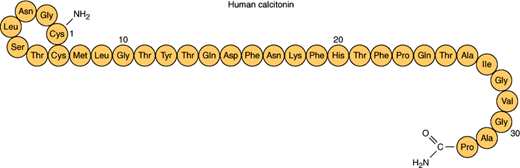
Calcitonin is a 32-amino-acid peptide hormone with a seven-member amino terminal disulfide ring and carboxyl terminal prolineamide (Figure 17–14). Differential processing of the calcitonin gene can lead to the production of either calcitonin in C cells or calcitonin gene-related peptide in neurons. Although both calcitonin and calcitonin gene-related peptide have demonstrated clinical effects in pharmacologic doses, the function of the peptides at normal physiologic levels is unknown. C-cell tumors may release both peptides.
Hypercalcemia stimulates the release of calcitonin through the activation of CaSRs in C cells. Substantial changes in serum calcium are normally required to modulate the release of calcitonin. It is not known whether small physiologic changes in serum calcium, which rapidly modulate PTH secretion, elicit significant changes in calcitonin levels. The GI hormones cholecystokinin and gastrin are also secretagogues for calcitonin.
Calcitonin secretion in vivo is assessed by measuring serum levels with a two-site radioimmunoassay.
Calcitonin interacts with receptors in kidney and bone. This interaction stimulates adenylyl cyclase activity and the generation of cAMP (as shown in Figure 17–5 for PTH). In the kidney, receptors for calcitonin are localized in the cortical ascending limb of Henle loop, whereas in bone calcitonin receptors are found on osteoclasts.
The main function of calcitonin is to lower serum calcium, and this hormone is rapidly released in response to hypercalcemia. Calcitonin inhibits osteoclastic bone resorption and rapidly blocks the release of calcium and phosphate from bone. The latter effect is apparent within minutes after the administration of calcitonin. These effects ultimately lead to a fall in serum calcium and phosphate.
Calcitonin acts directly on osteoclasts and blocks the resorption of bone induced by hormones like PTH and vitamin D. The potency of calcitonin depends on the underlying rate of bone resorption. Calcitonin also has a modest effect on the kidney to produce mild phosphaturia. With continued administration of calcitonin, “escape” from its effects on serum calcium occurs.
The overall importance of calcitonin in the maintenance of calcium homeostasis is unclear. Serum calcium concentrations are normal in patients after thyroidectomy, which removes all functioning C cells. Similarly, calcitonin typically rises to very high levels in patients with medullary carcinoma of the thyroid with no apparent effect on serum calcium levels.
Checkpoint
What are the actions of calcitonin?
What is the effect of thyroidectomy on serum calcium?
Pathophysiology of Selected Disorders of Calcium Metabolism
Primary hyperparathyroidism is due to excessive production and release of PTH by the parathyroid glands. The prevalence of hyperparathyroidism is approximately 1:1000 in the United States, and the incidence of the disease increases with age. The patient group most frequently affected is postmenopausal women.
Primary hyperparathyroidism may be caused by any of the following: adenoma, hyperplasia, or carcinoma (Table 17–1). Chief cell adenomas are the most common cause, accounting for almost 85% of all cases. The vast majority of parathyroid adenomas occur sporadically and are solitary.
Parathyroid hyperplasia refers to an enlargement or abnormality of all four glands. In atypical forms of hyperplasia, only one gland may be enlarged, but the other three glands typically show at least slight microscopic abnormalities such as increased cellularity and reduced fat content. The distinction between hyperplasia and multiple adenomas is challenging and usually requires the examination of all four glands. Key characteristics for judging whether a gland is normal or not are its size, weight, and histologic features.
Parathyroid hyperplasia may be part of the autosomal dominant multiple endocrine neoplasia (MEN) syndromes (Table 17–2). In patients with MEN-1, caused by mutations in the MEN1 gene, which encodes the protein menin, there is high penetrance of hyperparathyroidism, affecting as many as 95% of patients. When their glands are examined microscopically, there are usually abnormalities in all four glands. Recurrent hyperparathyroidism, even after initially successful surgery, is common in these patients. Hyperparathyroidism also occurs in MEN-2A, although at a much lower frequency (about 20%). Familial hyperparathyroidism, without other features of MEN syndromes, characteristically involves all four glands, but there is often asynchrony in the presentation of the hyperparathyroidism. Kindreds with isolated hyperparathyroidism and mutations in menin are considered to be allelic variants of MEN-1. The hyperparathyroidism-jaw tumor syndrome and familial isolated hyperparathyroidism are causes of autosomal dominant hyperparathyroidism. The former often includes ossifying fibromas of the jaw and renal tumors and is caused by inactivating germline mutations in the HRPT2 gene that encodes the protein parafibromin.
|
|
|
|
|
|
Parathyroid carcinoma is a rare malignancy, but the diagnosis should be considered in a patient with severe hypercalcemia and a palpable cervical mass. At surgery, cancers are firmer than adenomas and more likely to be attached to adjacent structures. It is sometimes difficult to distinguish parathyroid carcinomas from adenomas on histopathologic grounds. Vascular or capsular invasion by tumor cells is a good indicator of malignancy, but these features are not always present. In many cases, local recurrences or distant metastases to liver, lung, or bone are the clinical findings that support this diagnosis. Approximately 20% of patients with the hyperparathyroidism-jaw tumor syndrome and germline mutations in the HRPT2 gene (described above) develop parathyroid cancer. Furthermore, mutations in HRPT2 have also been found in familial isolated hyperparathyroidism and in sporadic parathyroid cancers. The normal cellular function of parafibromin is unknown.
Secondary hyperparathyroidism implies diffuse glandular hyperplasia resulting from a defect outside the parathyroids. Secondary hyperparathyroidism in patients with normal kidney function may be observed in patients with severe calcium and vitamin D deficiency states (see below). In patients with chronic kidney disease, there are many causative factors that contribute to the often dramatic enlargement of the parathyroid glands. These include decreased 1,25-(OH)2D production, reduced intestinal calcium absorption, skeletal resistance to PTH, and renal phosphate retention.
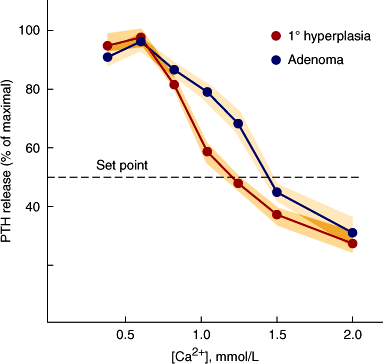
PTH secretion in primary hyperparathyroidism is excessive given the level of the serum calcium. At the cellular level, there is both increased cell mass and a secretory defect. The latter is characterized by reduced sensitivity of PTH secretion to suppression by the elevated serum calcium concentration. This qualitative regulatory defect is more common than truly autonomous secretion. Thus, parathyroid glands from patients with primary hyperparathyroidism are typically enlarged and, in vitro, demonstrate a “shift to the right” in their calcium setpoint for secretion (Figure 17–15). How these two defects interact in the pathogenesis of the disease remains to be fully elucidated.
Figure 17–15
PTH secretion in vitro from human parathyroid cells from patients with parathyroid adenomas and hyperplasia. The set-point for secretion is the calcium concentration at which PTH release is suppressed by 50%. This is shifted to the right in the majority of parathyroid adenomas compared to normal tissues, in which the set-point is approximately 1.0 mmol/L ionized calcium. (Redrawn, with permission, from Brown EM et al. Dispersed cells prepared from human parathyroid glands: distinct calcium sensitivity of adenomas vs primary hyperplasia. J Clin Endocrinol Metab. 1978;46:267.)
The genetic defects responsible for primary hyperparathyroidism have received considerable attention. Genes that regulate the cell cycle are thought to be important in the pathogenesis of a significant subset of parathyroid tumors. The PRAD1 gene (parathyroid rearrangement adenoma), whose product is a D1 cyclin, has been implicated in parathyroid tumor development and also in the pathogenesis of several malignant tumors (B-cell lymphomas, breast and lung cancers, and squamous cell cancers of the head and neck). Cyclins are cell cycle regulatory proteins. The PRAD1 gene is located on the long arm of chromosome 11, as is the gene encoding for PTH. Analysis of parathyroid tumor DNA suggests that a chromosome inversion event occurred, which led to juxtaposition of the 5-regulatory domain of the PTH gene upstream to the PRAD1 gene (Figure 17–16). Because regulatory sequences in the PTH gene are responsible for its cell-specific transcription, this inversion was initially postulated to lead to a parathyroid cell-specific overproduction of the PRAD1 gene product. Excessive cyclin would enhance the proliferative potential of the cells bearing this inversion and, given sufficient time, could induce PTH excess. A transgenic mouse model in which cyclin D1 is overexpressed in parathyroid tissue under the control of the PTH gene promoter provides proof for this pathogenetic mechanism of primary hyperparathyroidism.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


