Introduction
Insulin and glucagon, the two key hormones that orchestrate fuel storage and utilization, are produced by the islet cells in the pancreas. Islet cells are distributed in clusters throughout the exocrine pancreas. Together, they comprise the endocrine pancreas. Diabetes mellitus, a heterogeneous disorder, is the most common disease of the endocrine pancreas. Affecting 8% of the world’s adult population in 2011, the prevalence of diabetes continues to increase worldwide, having already more than doubled over the past 3 decades. Pancreatic tumors secreting excessive amounts of specific islet cell hormones are far less common, but their clinical presentations underscore the important regulatory roles of each hormone.
Normal Structure & Function of the Pancreatic Islets
The endocrine pancreas is composed of nests of cells (islets of Langerhans) that are distributed throughout the exocrine pancreas. This anatomic feature allows for their enzymatic isolation from the exocrine pancreas for islet cell transplantation. Although numbering in the millions, the multicellular islets comprise only 1% of the total pancreas. The endocrine pancreas has great reserve capacity; more than 70% of the insulin-secreting β cells must be lost before dysfunction occurs. Each of the four major islet cell types produces a different secretory product. Insulin-secreting β cells are the predominant cell type (60%). The majority of the remaining islet cells, glucagon-secreting α cells (30%) and somatostatin-secreting δ cells (<10%), secrete hormones that counter the effects of insulin. A fourth islet cell type, the pancreatic polypeptide (PP)–secreting cell (<1%), is primarily located in the posterior lobe of the head of the pancreas, an embryologically distinct region receiving a different blood supply.
The islets are much more highly vascularized than the exocrine pancreatic tissues (Chapter 15), with at least one major arteriole supplying each islet. The majority of islet cells are closely apposed to the vasculature and to islet cells of opposing types, suggesting an important role for endocrine (via the microcirculation) and/or intra-islet paracrine (via the interstitium) effects on hormone release (Figure 18–1). Blood from the islets then drains into the hepatic portal vein. Thus, the islet cell hormones pass directly into the liver, a major site of action of glucagon and insulin, before proceeding into the systemic circulation, allowing for much higher hepatic than systemic levels of pancreatic hormones.
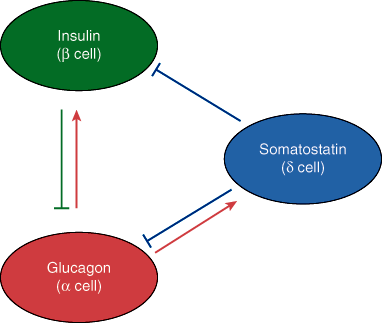
The islets are also abundantly innervated. Both parasympathetic and sympathetic axons enter the islets and either directly contact cells or terminate in the interstitial space between the cells. Neural regulation of islet cell hormone release, both directly through the sympathetic fibers and indirectly through stimulation of catecholamine release by the adrenal medulla, plays a key role in glucose homeostasis during stress.
Checkpoint
1. What percentage of islets must be lost before endocrine pancreatic dysfunction becomes manifest?
2. Identify the major hormone-secreting cells in an islet of Langerhans.
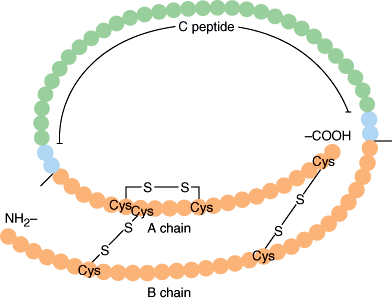
Insulin is a protein composed of two peptide chains (A and B chains) connected by two disulfide bonds (Figure 18–2). The precursor of insulin, preproinsulin (MW 11,500), is synthesized in the ribosomes and enters the endoplasmic reticulum of β cells, where it is promptly cleaved by microsomal enzymes to form proinsulin (MW 9000). Proinsulin, consisting of A and B chains joined by a 31-amino-acid C peptide, is transported to the Golgi apparatus, where it is packaged into secretory vesicles. While in the secretory vesicle, proinsulin is cleaved at two sites to form insulin (51 amino acids; MW 5808) and the C peptide fragment (Figure 18–2). Secretion of insulin is, therefore, accompanied by an equimolar secretion of C peptide and also by small amounts of proinsulin that escape cleavage. In the acidic environment of the secretory granules, stored insulin forms a hexamer in association with zinc atoms, dissociating into active monomers upon secretion. Insulin has a circulatory half-life of 3–5 minutes and is catabolized in both the liver and the kidney. Approximately 50% of insulin is catabolized on its first pass through the liver after it is secreted from the pancreas into the portal vein. In contrast, both C peptide and proinsulin are catabolized only by the kidney and, therefore, have half-lives three to four times longer than that of insulin itself. Recombinant human insulin or related analogs, which either enhance monomer formation (rapid acting) or decrease solubility (longer acting), are used clinically to treat diabetes.
Glucose is the primary physiologic stimulant of insulin release (Figure 18–3). Glucose entry into β cells is facilitated by one or more glucose transporters (GLUT-1, GLUT-2, and/or GLUT-3), which are in excess to glucose and allow for the bidirectional transport of glucose, thereby creating an equilibrium between extracellular and intracellular glucose concentrations. Once in the cell, the metabolism of glucose—rather than glucose itself—stimulates insulin secretion.
Figure 18–3
Schematic diagram of glucose-stimulated insulin release from β cell. Glucose enters the β cell via GLUT–mediated diffusion. Metabolism of glucose, the first step of which is controlled by glucokinase, results in ATP production. Cytosolic ATP, sensed by the sulfonylurea receptor subunit (SUR1) of ATP-dependent K+ channels (KATP), blocks the KATP channels and thus K+ efflux, resulting in cell depolarization. This allows Ca2+ to enter via voltage-dependent calcium channels, stimulating the exocytosis of insulin-containing secretory granules.
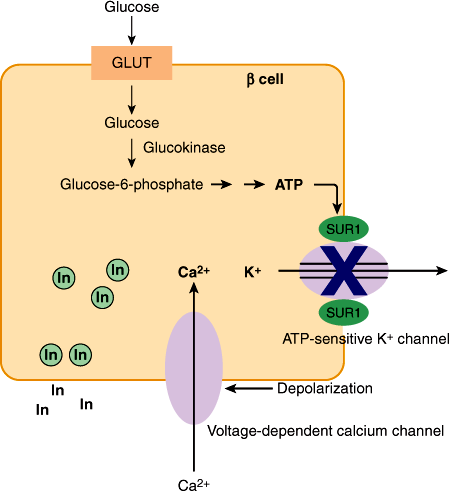
Glucokinase, an enzyme with low affinity for glucose whose activity is regulated by glucose, controls the first and rate-limiting step in glucose metabolism—the phosphorylation of glucose to form glucose 6-phosphate. This enzyme, by determining the rate of glycolysis, is thought to function as the β-cell glucose sensor. Glycolysis produces an increase in adenosine triphosphate (ATP), which is sensed by the sulfonylurea receptor subunit of ATP-dependent K+ channels (KATP) in the β-cell membrane, resulting in a closure of the channel. The resultant cell depolarization allows Ca2+ to enter, triggering exocytosis of insulin-containing granules. Sulfonylurea drugs used to treat type 2 diabetes stimulate insulin secretion in a glucose-independent fashion by binding to the sulfonylurea receptor subunit and blocking KATP.
Although glucose is the most potent stimulator of insulin release, other factors such as amino acids ingested with a meal or vagal stimulation also result in insulin release (Table 18–1). Up to 50% of insulin secretion in response to an oral glucose load can be attributed to enteric hormones (incretins) such as glucagon-like peptide-1 (GLP-1), which are released following oral ingestion of nutrients and enhance glucose-stimulated insulin secretion in β cells via activation of cAMP/PKA signaling pathways following binding to their G protein–coupled receptors. Glucagon similarly enhances glucose-stimulated insulin secretion, a counter-regulatory effect that allows for insulin-mediated disposal of glucose following the hepatic production of glucose induced by glucagon. Insulin secretion is inhibited by catecholamines and by somatostatin.
| β-Cell Insulin Release | δ-Cell Somatostatin Release | α-Cell Glucagon Release | |
|---|---|---|---|
| Nutrients | |||
| Glucose | ↑ | ↑ | ↓ |
| Amino acids | ↑ | ↑ | ↑ |
| Fatty acids | — | — | ↓ |
| Ketones | — | — | ↓ |
| Hormones | |||
| Enteric hormones | |||
| GLIP-1 | ↑ | ↓ | ↓ |
| GIP | ↑ | ↓ | ↑ |
| Insulin | ↓ | ↓? | ↓ |
| GABA | — | ↓ | ↓ |
| Somatostatin | ↓ | ↓ | ↓ |
| Glucagon | ↑ | ↑ | — |
| Cortisol | — | — | ↑ |
| Catecholamines | ↓ (α-adrenergic) | — | ↑ (β-adrenergic) |
| Neural | |||
| Vagal | ↑ | — | ↑ |
| α-Adrenergic | ↑ | — | ↑ |
| β-Adrenergic | ↓ | — | ↓ |
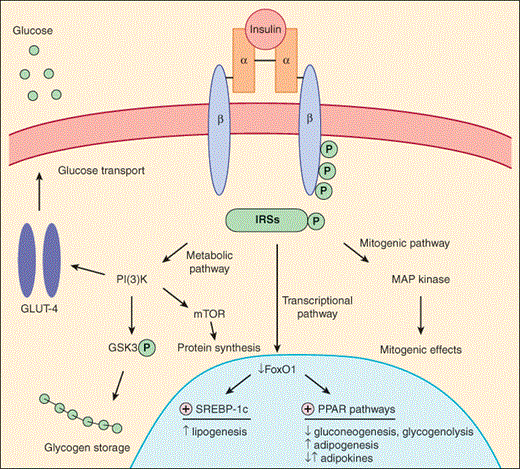
Insulin exerts its effects by binding to insulin receptors present on the surfaces of target cells (Figure 18–4). Insulin receptors are present in liver, muscle, and fat, the classic insulin-sensitive tissues responsible for fuel homeostasis. In addition, insulin can mediate other effects in nonclassic target tissues, such as the ovary, via interaction with insulin receptors or by cross-reactivity with insulin-like growth factor-1 (IGF-1) receptors. Binding of insulin to its receptor causes activation of a tyrosine kinase region of the receptor and autophosphorylation of the receptor. Activation of the insulin receptor initiates a phosphorylation cascade within the cell, beginning with the phosphorylation of a network of docking proteins (insulin receptor substrates [IRSs]) that engage and amplify downstream signaling molecules, ultimately leading to the biologic effects of insulin (eg, translocation of GLUT-4 glucose transporter to the plasma membranes of muscle and fat cells and activation of hepatic glycogen synthase).
Figure 18–4
Model of insulin receptor signaling. The insulin receptor is composed of two α and two β subunits linked by disulfide bonds. Binding of insulin to the extracellular α subunits activates a tyrosine kinase present in the cytoplasmic domain of the β subunit, resulting in autophosphorylation of the β subunit. Receptor kinase activation is also the critical first step in a cascade of intracellular events that begins with phosphorylation of multiple docking proteins (insulin receptor substrates [IRSs]). Once activated, these multifunctional proteins initiate complex intracellular signaling pathways. Binding of IRS to phosphatidylinositol 3-kinase (PI3-K) initiates a metabolic pathway, stimulating glucose uptake by translocation of the glucose transporter, GLUT-4, to the cell surface in skeletal muscle and adipose; stimulating glucose storage by the inactivation (via phosphorylation) of glycogen synthase kinase 3 (GSK3) and subsequent dephosphorylation and activation of glycogen synthase; and increasing protein synthesis via activation of the serine/threonine protein kinase, mechanistic target of rapamycin (mTOR). In contrast, mitogenic effects of insulin are mediated by a mitogen activated protein kinase (MAP) kinase pathway. Additionally, important transcriptional effects occur, many of which involve inactivation (via phosphorylation) of the transcription factor, FoxO1, which is abundant in insulin-sensitive tissues. This change, in concert with effects of other transcription factors (SREBP-1c, PPARs), allows for additional insulin-mediated effects, including increased lipogenesis vs. decreased gluconeogenesis and glycogenolysis in the liver, together with increased adipogenesis and lipid storage in adipose tissue.
Insulin plays a major role in fuel homeostasis (Table 18–2). Insulin mediates changes in fuel metabolism through its effects on three main tissues: liver, muscle, and fat. In these tissues, insulin promotes fuel storage (anabolism) and prevents the breakdown and release of fuel that has already been stored (catabolism). The total lack of insulin is incompatible with life, and the same is true of excess insulin.
| Insulin | Somatostatin | Glucagon | Catecholamines | Cortisol | Growth Hormone | |
|---|---|---|---|---|---|---|
| Liver | ||||||
| Fuel storage | ||||||
| ↑ | ↓ | ||||
| ↑ | ↓ | ||||
| Fuel breakdown | ||||||
| ↓ | ↑ | ↑ | ↑ | ||
| ↓ | ↑ | ↑ | ↑ | ↑ | |
| ↓ | ↑ | ||||
| Kidneys | ||||||
| Fuel breakdown | ||||||
| ↓ | ↑ | ||||
| Muscle | ||||||
| Fuel storage | ||||||
| ↑ | ↓ | ↓ | ↓ | ||
| Fuel breakdown | ||||||
| ↓ | ↑ | ||||
| Adipose tissue | ||||||
| Fuel storage | ||||||
| ↑ | |||||
| ↑ | |||||
| Fuel breakdown | ||||||
| ↓ | ↑ | ↑ | ↑ | ||
| Pancreas | ||||||
| Secretion of: | ||||||
| ↓ | ↓ | ↑ | ↓ | ||
| ↓ | ↓ | ↑ | ↑ | ↑ | |
| (↓?) | ↓ | ↑ | |||
In the liver, insulin promotes fuel storage by stimulation of glycogen synthesis and storage. Insulin inhibits hepatic glucose output by inhibiting gluconeogenesis (glucose synthesis) and glycogenolysis (glycogen breakdown). By also stimulating glycolysis (metabolism of glucose to pyruvate), insulin promotes the formation of precursors for fatty acid synthesis. Insulin stimulates lipogenesis (biosynthesis of fatty acids from glucose) while inhibiting fatty acid oxidation and the production of ketone bodies (ketogenesis), an alternative fuel produced only in the liver that can be used by the brain when glucose is not available.
Although hepatic uptake of glucose, occurring via low-affinity GLUT-2 transporters, is not regulated by insulin, glucose uptake both in muscle and in fat is regulated by insulin, which causes the rapid translocation of an insulin-sensitive glucose transporter (GLUT-4) to the surface of these cells. Uptake of glucose by muscle accounts for the vast majority (85%) of insulin-stimulated glucose disposal. In muscle, insulin promotes the storage of glucose by stimulating glycogen synthesis and inhibiting glycogen catabolism. Insulin also stimulates protein synthesis in muscle.
Insulin stimulates fat storage in adipose tissue by stimulating lipoprotein lipase, the enzyme that hydrolyzes the triglycerides carried in very-low-density lipoproteins (VLDLs) and other triglyceride-rich lipoproteins to fatty acids, which can then be taken up by fat cells. Increased glucose uptake caused by upregulation of the GLUT-4 transporter also aids in fat storage because this increases the levels of glycerol phosphate, a substrate in the esterification of free fatty acids, which are then stored as triglycerides. In fat cells, insulin also inhibits lipolysis, preventing the release of fatty acids, which are potential substrates for hepatic ketone body synthesis and/or hepatic VLDL-triglyceride synthesis. Insulin exerts this effect by preventing phosphorylation of hormone-sensitive lipase, thus inactivating the enzyme that hydrolyzes stored triglycerides to releasable fatty acids. Together, these changes result in increased fat storage in adipose tissue.
Checkpoint
3. What is the half-life of insulin? How is it catabolized? What percentage is extracted on first pass through the liver?
4. How do the half-lives of C peptide and proinsulin compare with that of insulin?
5. List the main substances that stimulate insulin secretion.
6. What characteristics of the β-cell glucose transporter allow intracellular glucose levels to equal those of the extracellular space?
7. What is the probable “glucose sensor” in the β cell?
8. What are the major inhibitors of insulin secretion?
9. What are the current thoughts on the mechanisms of insulin action?
10. Which tissues are insulin dependent for glucose uptake?
11. What are three ways in which insulin stimulates fat storage?
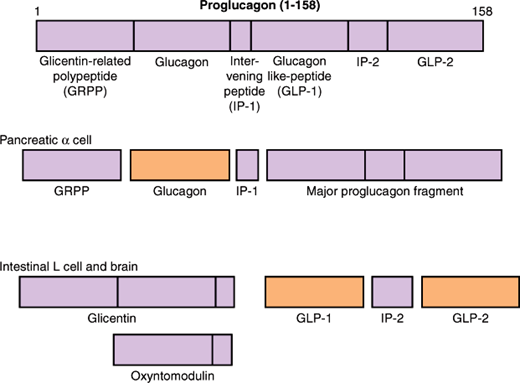
Glucagon, a 29-amino-acid peptide, is produced in α cells of the pancreas by the proteolytic processing of proglucagon, a larger precursor protein. In addition to the pancreas, proglucagon is also expressed in the intestine and brain. While glucagon is the major bioactive metabolite produced in the pancreatic α cell, differential processing by L cells in the intestine results in the production of glucagon-like peptide (GLP)-1 and GLP-2 in response to a meal (Figure 18–5). This tissue-specific processing results in peptides with opposing effects on carbohydrate metabolism; pancreatic glucagon opposes the hepatic effects of insulin, whereas GLPs acts as incretins, gut-derived peptides that enhance glucose-stimulated insulin secretion. The circulatory half-life of glucagon is 3–6 minutes. Like insulin, glucagon is metabolized in the liver and kidneys, with kidneys, rather than liver, playing a significant role. Long-acting analogs of GLP-1, which also stimulate β-cell proliferation and increase β-cell mass, or enzyme inhibitors that extend the half-life of endogenous GLIP-1 are a newer and important class of drugs used for the treatment of type 2 diabetes mellitus.
In contrast to the stimulation of insulin secretion by glucose, glucagon secretion is inhibited by glucose (Table 18–1). However, the relative importance of direct sensing of glucose by the α cell vs. indirect paracrine/endocrine effects of other pancreatic factors in regulating glucagon secretion is a matter of debate. Current evidence suggests that insulin plays a major role in modulating (ie, inhibiting) glucagon secretion. Moreover, the loss of the suppressive effects of increased pancreatic insulin in diabetes in response to hyperglycemia results in an inappropriately high level of glucagon, which contributes to the hyperglycemia of diabetes mellitus. Other pancreatic factors inhibiting glucagon secretion include somatostatin and two additional β-cell secretory products, γ-aminobutyric acid (GABA), and insulin-associated zinc. Like insulin, glucagon secretion is stimulated by amino acids, an important regulatory feature in the metabolism of protein meals. In contrast, fatty acids and ketones inhibit glucagon secretion. Other counter-regulatory hormones such as catecholamines (via a predominating β-adrenergic effect) and cortisol stimulate glucagon release.
The major biological role of glucagon is to maintain normal glucose levels during fasting by inducing hepatic glucose production, thus counteracting the hepatic effects of insulin. Therefore, the liver is the major target organ for glucagon action. Glucagon binds to a G protein–coupled glucagon receptor present on the cell surface of hepatocytes, activating adenylyl cyclase and generating cAMP. Cyclic AMP activates protein kinase A, which activates gene transcription for the enzymes responsible for the biologic activity of glucagon in the liver, and subsequently phosphorylates and activates these same enzymes. There is also some evidence that the glucagon receptor may act via an adenylyl cyclase-independent mechanism by stimulation of phospholipase C.
The actions of glucagon were first demonstrated in 1921 by Banting and Best when they observed a mild transient hyperglycemia preceding insulin-induced hypoglycemia when testing pancreatic extracts in vivo. Glucagon is a counter-regulatory hormone, acting in a catabolic fashion to oppose the effects of insulin. Indeed, glucagon injections are used clinically to treat severe hypoglycemia. Hepatic effects of glucagon (Table 18–2) include the following: (1) increased hepatic glucose output via the release of glycogen stores (glycogenolysis) and, in concert with other counter-regulatory hormones, stimulation of hepatic glucose synthesis (gluconeogenesis); (2) increased hepatic uptake of amino acids, which fuels gluconeogenesis; and (3) stimulation of fatty acid oxidation and ketogenesis, thus providing an alternative fuel (ketone bodies) that can be used by the brain when glucose is not available. The physiologic significance of glucagon receptors in non-hepatic tissue (kidney, adipose, pancreas) is less certain. For example, glucagon, while less potent, shares with GLIP-1 the ability to enhance glucose-induced β-cell insulin secretion.
Like preproglucagon, preprosomatostatin is synthesized in the pancreas, GI tract, and brain, where it is differentially processed in a tissue-specific fashion to produce several biologically active peptides. Somatostatin-14 (SS-14), the first somatostatin to be isolated, is a 14-amino-acid peptide that was initially discovered in the hypothalamus as the factor responsible for the inhibition of growth hormone release. Only later was it appreciated that δ cells of the pancreas also secrete SS-14. In brain and intestine, somatostatin-28 (SS-28), an amino-terminally extended peptide that includes the 14-amino-acid sequence of SS-14, is also produced from preprosomatostatin and has a range of action comparable to that of SS-14 but a potency that is somewhat greater. The half-life of somatostatin (<3 minutes) is shorter than that of insulin or glucagon. Because somatostatin has been shown to inhibit the synthesis and secretion of most peptide hormones, synthetic somatostatin analogs, such as octreotide, that have a much longer half-life (hours) have been developed for clinical use in inhibiting ectopic peptide hormone production by a variety of tumors. The same secretagogues that stimulate insulin secretion also stimulate somatostatin (Table 18–1). These include glucose, amino acids, enteric hormones, and glucagon.
Somatostatin exerts its effects via binding to a family of inhibitory G (Gi) protein–coupled receptors (SST1-5) that are distributed in a tissue-specific fashion. In all tissues where somatostatin is produced, it acts primarily in an inhibitory fashion. In the endocrine pancreas, somatostatin is thought to act via paracrine effects on the other islet cells, inhibiting the release of insulin and glucagon (Table 18–1) and of PP. In addition, somatostatin acts in an autocrine fashion to inhibit its own release. In the GI tract, somatostatin retards the absorption of nutrients through multiple mechanisms, including the inhibition of gut motility, inhibition of several enteric peptides, and inhibition of pancreatic exocrine function. Consistent with the multiple inhibitory effects of this peptide, the synthetic somatostatin analog octreotide has multiple clinical uses, including inhibition of hormone production by pituitary adenomas, inhibition of certain types of chronic diarrhea, inhibition of tumor growth, and inhibition of bleeding from esophageal varices.
Pancreatic peptide (PP), a 36-amino-acid peptide is produced by the PP cells (F cells) in the islets of the posterior lobe of the head of the pancreas, is released in response to a mixed meal, an effect that appears to be mediated by protein and vagal stimulation. While long known to inhibit gastrointestinal motility and pancreatic exocrine secretions, more recent evidence suggests that PP may also control satiety and weight, inhibiting food intake and stimulating energy expenditure. These latter effects of PP (a member of the neuropeptide Y family of peptide hormones) are mediated centrally, via binding to an inhibitory G protein–coupled Y4 receptor and are thought to involve inhibition of hepatic vagal nerve afferent activity.
Checkpoint
12. What are some important stimulators and inhibitors of glucagon secretion?
13. What is the major target organ for glucagon? What are the mechanisms of glucagon action?
14. What metabolic pathways are sensitive to glucagon, and how are they affected?
15. What hormone antagonizes glucagon’s effect on metabolic pathways?
16. Where else in the body besides the islets of Langerhans is glucagon made?
17. By what mechanisms can GLPs enhance glucose-stimulated insulin secretion?
18. What is the role of somatostatin in the islets of Langerhans?
Carbohydrate metabolism is primarily controlled by the relative amounts of insulin and glucagon produced by the endocrine pancreas (Table 18–2; Figure 18–6). Conversely, dysregulation of both of these hormones contributes to hyperglycemia in diabetes. Under normal conditions, when plasma glucose levels are high, the actions of insulin predominate, including insulin suppression of glucagon secretion. Fuel storage is promoted by insulin stimulation of glycogen storage in the liver; glucose uptake, glycogen synthesis, and protein synthesis by muscle; and fat storage by adipose tissue. Insulin inhibits the mobilization of substrates from peripheral tissues and opposes any effects of glucagon on the stimulation of hepatic glucose output.
Figure 18–6
Mean rates of insulin and glucagon delivery from an artificial pancreas at various blood glucose levels. The device was programmed to establish and maintain normal blood glucose in nine patients with type 1 DM. The values for hormone output approximate the output of the normal human pancreas. The shape of the insulin curve also resembles the insulin response of incubated β cells to graded concentrations of glucose. (Copyright © 1977 American Diabetes Association. Marliss EB et al. Normalization of glycemia in diabetics during meals with insulin and glucagon delivery by the artificial pancreas. Diabetes. 1977;26:663–72. Reprinted, with permission, from the American Diabetes Association.)
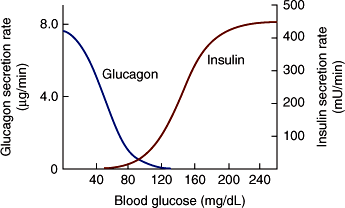
In contrast, when glucose levels are low, plasma insulin levels are suppressed and the effects of glucagon predominate in the liver (ie, increased hepatic glucose output and ketone body formation). In the absence of insulin, muscle glucose uptake is markedly decreased, muscle protein is catabolized, and fat is mobilized from adipose tissue. Therefore, with insulinopenia, glucose loads cannot be cleared, and substrates for hepatic gluconeogenesis (amino acids, glycerol) and ketogenesis (fatty acids)—processes that are stimulated by glucagon—are increased.
After an overnight fast, the liver plays a primary role in maintaining blood glucose by producing glucose at the same rate at which it is used by resting tissues. Glucose uptake and utilization occur predominantly in tissues that do not require insulin for glucose uptake, such as the brain. Hepatic glucose output is stimulated by glucagon and is primarily due to glycogenolysis, which can provide, on average, an 8-hour supply of glucose. The low levels of insulin that are present (basal secretion of 0.25–1.0 unit/h) are insufficient to block the release of fatty acids from fat, which provide fuel for muscles (fatty acid oxidation) and substrate for hepatic ketogenesis. However, these levels of insulin are sufficient to prevent excessive lipolysis, ketogenesis, and gluconeogenesis, thus preventing hyperglycemia and ketoacidosis.
With prolonged fasting (>24–60 hours), liver glycogen stores are depleted. Glucagon levels rise slightly, and insulin levels decline further. Gluconeogenesis now becomes the sole source of hepatic glucose production, using substrates such as amino acids that are mobilized from the periphery at a greater rate. With starvation, a switch occurs in the liver from gluconeogenesis to the production of ketones, an alternative fuel source that provides 90% of the energy used by the brain, a critical organ that accounts for 25% of basal metabolic energy needs. In this manner, survival is prolonged as muscle protein is conserved in favor of increased mobilization of fatty acids from adipose tissue, a process made possible by increased insulinopenia. The liver then converts fatty acids to ketone bodies, a process that is stimulated by glucagon. With prolonged fasting or starvation, the kidney also begins to contribute significantly to gluconeogenesis.
With ingestion of a carbohydrate load, insulin secretion is stimulated and glucagon is suppressed. Hepatic glucose production and ketogenesis are suppressed by the high ratio of insulin to glucagon. Insulin stimulates hepatic glycogen storage. Insulin-mediated glucose uptake, which occurs primarily in muscle, is also stimulated, as is muscle glycogen synthesis. Fat storage occurs in adipose tissue.
With ingestion of a protein meal, both insulin and glucagon are stimulated. In this way, insulin stimulates amino acid uptake and protein formation by muscle. However, stimulation of hepatic glucose output by glucagon counterbalances the tendency of insulin to cause hypoglycemia.
During severe stress, when fuel delivery to the brain is in jeopardy, counter-regulatory hormones, in addition to glucagon, act synergistically. They maintain blood glucose levels by maximizing hepatic output of glucose and peripheral mobilization of substrates and by minimizing fuel storage. Glucagon and epinephrine act within minutes to elevate blood glucose, whereas the counter-regulatory effects of cortisol and growth hormone are not seen for several hours. Epinephrine, cortisol, and growth hormone stimulate glucagon release, whereas epinephrine inhibits insulin, thus maximally increasing the glucagon-insulin ratio. In addition, these three hormones act directly on the liver to increase hepatic glucose production and peripherally to stimulate lipolysis and inhibit insulin-sensitive glucose uptake. During severe stress, hyperglycemia may actually result from the combined effects of counter-regulatory hormones.
Similar but less marked effects occur in response to exercise when glucagon, catecholamines, and, to a lesser extent, cortisol help meet the several-fold increase in glucose utilization rates due to exercising muscle by increasing hepatic glucose output and lipolysis of fat stores, effects that are made possible by a lowering of insulin levels. Low insulin levels also allow muscles to use glycogen stores for energy.
Kidney and liver both express the enzymes required to augment the glucose pool by gluconeogenesis and the secretion of glucose stored as glycogen. While the kidney contributes little to the glucose pool during an overnight fasting, it contributes approximately 50% of endogenous glucose production during a prolonged (>40 hours) fast. Gluconeogenesis predominates in the kidney as its glycogen stores are minimal, a process that is stimulated by epinephrine, inhibited by insulin, and unaffected by glucagon.
Checkpoint
19. In insulinopenic states, why are substrates for hepatic gluconeogenesis and ketogenesis increased?
20. What is the effect of a protein meal on insulin versus glucagon secretion?
21. What is the difference in time course of action of the various counter-regulatory hormones?
Pathophysiology of Selected Endocrine Pancreatic Disorders
Diabetes mellitus is a heterogeneous disorder defined by the presence of hyperglycemia. Diagnostic criteria for diabetes include the following: (1) a fasting plasma glucose of 126 mg/dL or more, (2) classic symptoms of hyperglycemia plus a random plasma glucose of 200 mg/dL or more, or (3) a plasma glucose level of 200 mg/dL or more after an oral dose of 75 g of glucose (oral glucose tolerance test, OGTT). More recently, following the establishment of standardized assays, glycated hemoglobin (HbA1C), which correlates with chronic increases in glucose, has been used to diagnose diabetes when HbA1C levels 6.5% or more are documented using an appropriate methodology.
Hyperglycemia in all cases is due to a functional deficiency of insulin action. Deficient insulin action can be due to a decrease in insulin secretion by the β cells of the pancreas, a decreased response to insulin by target tissues (insulin resistance), or an increase in the counter-regulatory hormones that oppose the effects of insulin. The relative contributions of these three factors form the basis for the classification of this disorder into subtypes and also helps to explain the characteristic clinical presentations of each subtype (Table 18–3).
|
Diabetes prevalence worldwide, which has been increasing over the past few decades, reached 8% in 2011 in those 20 years or older (and a prevalence of 11% in the United States). More than 90% of cases of diabetes mellitus are believed to occur in the context of a genetic predisposition and are classified as either type 1 diabetes mellitus (DM) or type 2 DM (Tables 18–3 and 18–4). Type 1 DM is much less common than type 2 DM, accounting for 5–10% of cases of primary diabetes. Type 1 DM is characterized by autoimmune destruction of pancreatic β cells with resultant severe insulin deficiency. In a minority of patients, the cause of type 1 DM is unknown. The disease commonly affects individuals younger than 30 years; a bimodal peak in incidence occurs around age 5–7 years and at puberty. Although autoimmune destruction of the β cells does not occur acutely, clinical symptoms usually do. Patients present after only days or weeks of polyuria, polydipsia, and weight loss with markedly elevated serum glucose concentrations. Ketone bodies are also increased because of the marked lack of insulin, resulting in severe, life-threatening acidosis (diabetic ketoacidosis). Patients with type 1 DM require treatment with insulin.
| Type 1 DM | Type 2 DM | |
|---|---|---|
| Epidemiology | ||
| Age at diagnosis | Childhood | Adult |
| (incidence increasing with obesity in children) | ||
| Prevalence (in USA) | 0.2%, age <20 years | 11%, age >20 years |
| Phenotype | ||
| β-Cell insulin secretion abnormal | Absolute deficiency | Impaired secretion |
| Insulin resistance | No | Yes |
| Obese | No | Yes |
| BMI | Usually <25 | >25 in 85%; >30 in 50% |
| Autoimmune disease | Yes | No |
| In 90% | |
| Postulated environmental triggers | Viral infections, dietary exposures (cow’s milk, cereal) | Obesity (diet, exercise) |
| Genotype | ||
| Concordance in monozygotic twins | <50% | >70% |
| Incidence in offspring | ||
| 2–5% | 15% |
| 10% | 50% |
| Genetic loci associated with risk | HLA class II genes | Heterogeneous sets of interacting genes |
Type 2 DM differs from type 1 DM in several distinct ways (Table 18–4): It accounts for the overwhelming majority of diabetes (90–95%); has a stronger genetic component; occurs most commonly in adults; increases in prevalence with age (ie, 18% of individuals older than 65 years worldwide, or 27% in the United States); occurs more commonly in Native American, Mexican American, and African American populations in the United States; and is associated with increased resistance to the effects of insulin at its sites of action as well as a decrease in insulin secretion by the pancreas. It is often (85% of cases) associated with obesity, an additional factor that increases insulin resistance. Thus, the rising prevalence of diabetes worldwide has been associated with an increasing prevalence of obesity (12%). Insulin resistance is the hallmark of type 2 DM. Because these patients often have varying amounts of residual insulin secretion that prevent severe hyperglycemia or ketosis, they often are asymptomatic and are diagnosed 5–7 years after the actual onset of disease (frank hyperglycemia) by the discovery of an elevated fasting glucose on routine screening tests. Population screening surveys show that a remarkable 30% of cases of type 2 DM in the United States, or 50% of cases worldwide, remain undiagnosed. Additionally, it is estimated that one-third of the adult population in the United States is insulin-resistant and hence in a pre-diabetic (normoglycemic) state. Once diagnosed with type 2 DM, most individuals (70%) are managed with lifestyle modification (eg, diet, exercise, weight management) alone or in combination with medications that (1) enhance endogenous glucose-independent insulin secretion (sulfonylureas), (2) amplify endogenous glucose-dependent insulin secretion (incretins, such as GLP-1), (3) decrease insulin resistance in hepatic or peripheral tissues (eg, metformin or glitazones, respectively), or (4) interfere with intestinal absorption of carbohydrates (eg, intestinal α-glycosidase inhibitors). A new class of drugs inhibiting the transporter responsible for renal glucose reabsorption (sodium-glucose co-transporter 2 [SGLT2]) is also being developed for use in type 2 DM. Type 2 diabetic patients do not usually require insulin treatment for survival. However, some patients with advanced type 2 DM are treated with insulin to achieve optimal glucose control.
An epidemic of type 2 DM is occurring worldwide, particularly in non-European populations; it has been estimated that 1 in 3 children born after 2000 will develop diabetes, particularly type 2 DM, in their lifetime. Thus, while type 1 DM remains the most common cause of diabetes in children younger than 10 years (regardless of ethnicity) and in older, non-Hispanic white children, type 2 DM accounts for more than 50% of the diagnoses in older children of Hispanic, African American, Native American, and Asian Pacific Islander ancestry. In all age groups and ethnicities, this increased incidence of type 2 DM is associated with obesity.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


