CHAPTER 17 Disorders of phagocyte function
Introduction
Metchnikoff in the 19th century identified phagocytes as an essential and primary component in the elimination of a harmful agent, and predicted that defects in phagocyte function would predispose the host to microbial invasion.1 Subsequently, patients with decreased phagocyte counts or compromised phagocytic function (for either inherited or acquired reasons) have been shown to suffer recurrent and often fatal infections. The cells implicated in these disorders arise primarily from the myeloid lineage, namely eosinophils, neutrophils, monocytes, macrophages and dendritic cells. During an infection and the subsequent inflammatory reaction, phagocyte production and activation is initiated by enhanced proliferation and maturation of phagocyte precursors in the bone marrow (BM), which are then released into the circulation at an accelerated rate (Fig. 17.1A). Competent phagocytic cells (Fig. 17.1B) activate specific adhesive receptors which aid in rolling and firm adhesion to the endothelium. In response to gradients of chemokinetic and chemotactic molecules, the phagocytes traverse the vascular endothelial wall and migrate towards the site of infection. Interactions between the phagocyte and microbe precede engulfment, degranulation and activation of the respiratory burst, which in combination result in microbial killing primarily within the phagocytic vacuole. Defects in any part of this complex process manifest as immunodeficiency of phagocyte function, highlighted in Fig. 17.1. Primary defects of phagocytes are the result of genetic mutations (Table 17.1), many of which have been identified very recently, and the results of which affect adhesion, migration, respiratory burst activity and degranulation.
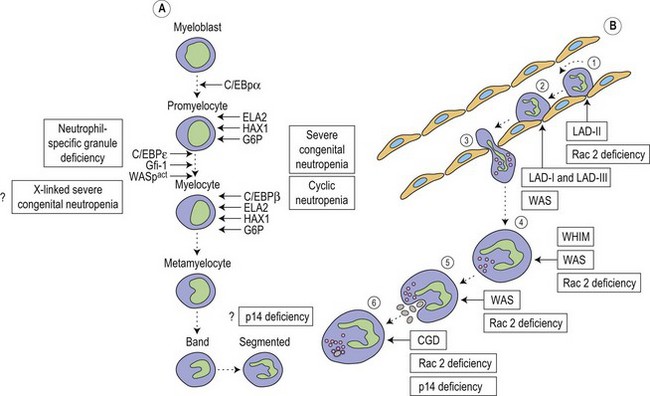
Table 17.1 Phagocyte immunodeficiency diseases
| Disease | Genetic defect | Phenotypic defect |
|---|---|---|
| Disorders of phagocyte maturation | ||
| Severe congenital neutropenia | AD or sporadic ELA2 mutation HAX1 mutation Gfi-1 mutation | Severe neutropenia. Developmental arrest of myeloid precursors usually at promyelocytic stage. Increased apoptosis |
| Cyclic neutropenia | AD or sporadic ELA2 mutation | Fluctuations between normal granulocyte counts and severe neutropenia with 21-day periodicity. Increased apoptosis in myeloid precursors |
| X-linked neutropenia | X-linked activating WAS mutation | Neutropenia and monocytopenia, and lymphocyte abnormalities |
| Neutrophil specific granule deficiency | AR C/EBPε mutation | Absent secondary and tertiary granule proteins. Primary granules lack defensins |
| Glucose-6-phosphatase deficiency | AR G6PC3 mutation | Loss of glucose-6-phosphatase activity. Increased susceptibility to apoptosis |
| p14 deficiency | AR p14 (MAPBPIP) mutation | Defective lysosome function. Neutropenia, hypoglammaglobulinemia, short stature and hypopigmentation |
| Deficiencies of adhesion and migration | ||
| LAD-I | AR ITGB2 mutation | High peripheral neutrophil counts and inability to make pus. Lack or reduction of CD18 expression |
| LAD-II | AR GDP-fucose transporter mutation | Granulocytes unable to bind to selectins on endothelium. Congenital disorder of fucosylation |
| LAD-III | AR RASGRP2 mutation FERMT3 mutation | High peripheral neutrophil counts and severe bleeding tendency. Defective integrin activation |
| WHIM | AD or AR CXCR4 mutation | Warts, hypoglammaglobulinemia, infections and myelokathexis |
| Phagocyte signalling abnormalities | ||
| WAS | X-linked WAS mutation | Dysfunctional actin polymerization in hematopoietic cells. Defects of cell activation, adhesion, migration and phagocytosis |
| Rac2 deficiency | AD Rac2 mutation | Abnormal granulocyte chemotaxis, respiratory burst and degranulation. High peripheral neutrophil counts |
| Deficiencies of the phagocyte respiratory burst | ||
| CGD | X-linked gp91phox mutation AR p47phox, p22phox, p40phox and p67phox mutations | Phagocytes unable to produce superoxide |
AD, autosomal dominant; AR, autosomal recessive; C/EBP, CGD, chronic granulomatous disease; CXCR4, CXC-containing chemokine receptor-4; CCAAT/enhancer binding protein; ELA2, elastase 2; FERMT3, fermitin family homolog 3; G6PC3, glucose-6-phosphate catalytic subunit 3; Gfi-1, growth factor independent-1 transcription repressor; HAX1, HCLS1-associated protein X-1; ITGB2, integrin beta-2; LAD, leukocyte adhesion deficiency; MAPBPIP, MAPBP-interacting protein; Rac2, Ras-related C3 botulinum toxin substrate 2; RASGRP2, RAS guanyl releasing protein-2; WAS, Wiskott–Aldrich syndrome; WHIM, warts, hypoglammaglobulinemia, infections and myelokathexis.
Disorders of phagocyte maturation
Severe congenital neutropenia (SCN) is a heterogeneous disease with different forms of inheritance. Patients present with life-threatening infections due to very low circulating neutrophil counts and are usually diagnosed at birth or soon thereafter.2 Although SCN was originally described as Kostmann syndrome,3 this form is now recognized as a discrete genetic disorder. Studies of BM of SCN patients indicate a relatively selective defect of neutrophil formation and a maturational arrest at the promyelocytic stage (Fig. 17.1A). Cyclic neutropenia, in contrast, is characterized by regular and consistent oscillations of neutrophil count, usually with a periodicity of 21 days, which clinically manifests as fever, mouth ulcers and infection at the nadir (which lasts from 3 to 6 days) of the neutrophil count.4,5 Over 90% of patients with either disorder respond to granulocyte-colony stimulating factor (G-CSF) therapy,6,7 although refractory cases and those complicated by hematologic malignancy may be rescued by hematopoietic stem cell transplantation. Initially approximately 10% of patients with SCN were thought to develop acute myeloid leukemia or myelodysplasia, often accompanied by monosomy 7 and cytogenetic abnormalities including trisomy 21.8–10 More recent data, however, from the Severe Chronic Neutropenia International Registry (http://depts.washington.edu/registry/), indicate that the incidence is significantly higher than previously recognized with a cumulative incidence of 21%.6,11,12 It is thought that an association exists between G-CSF therapy, acquired mutations of the G-CSF receptor and the risk of leukemia,6,10,13,14 but proving such an association is hampered by the fact that almost all patients are on G-CSF treatment.
Most frequently, SCN follows an autosomal dominant inheritance, or arises sporadically. Occasionally, SCN may arise as an autosomal recessive disorder, indicating that there is genetic heterogeneity. Similarly, cyclic neutropenia usually follows an autosomal dominant or sporadic pattern of inheritance. The underlying genetic lesions that account for the majority of cases of SCN or cyclic neutropenia have recently been identified. Detailed studies of families with autosomal dominant cyclic neutropenia identified a genetic lesion mapping to a region on chromosome 19p13.3 containing the genes for three neutrophil serine proteases, azurcidin, proteinase 3 and neutrophil elastase (encoded by the ELA2 gene). It was subsequently determined that mutations in ELA2 (which is synthesized at the promyelocytic stage of neutrophil development; Fig. 17.1A), were responsible for this form of cyclic neutropenia.15 Furthermore, because many of the hematological features of cyclic neutropenia are shared with SCN, it was later determined that heterozygous mutations in the same gene account for about 50% of autosomal dominant SCN.15–19 At least 52 different mutations have been described, but there does not seem to be a clear association between particular ELA2 mutations and the clinical form of neutropenia (cyclic vs. SCN), although the diversity of mutations in SCN appears to be wider than in cyclic neutropenia. The most common type of mutation in cyclic neutropenia disrupts the normal splice donor site at the end of the fourth exon, causing an in frame deletion of 10 amino acids and is seldom seen in SCN.20 In SCN many mutations are located in the fifth exon and it appears that the G185R mutation is associated with a particularly severe phenotype.16,20
Neutrophil elastase is a monomeric, 218-amino acid (25 kDa), chymotryptic serine protease synthesized by promyelocytes and promonocytes during the early stages of primary granule production, and is formed as a proenzyme during differentiation before final storage in azurophilic granules in its active state.21 It has activity against many proteins including matrix components and clotting factors, and is effectively neutralized by a number of endogenous inhibitors including the serpins α1-antitrypsin and monocyte/neutrophil elastase inhibitor (MNEI; gene symbol ELANH2) and the non-serpin, elfin. The mechanism by which elastase defects results in neutropenia remains uncertain. One possibility is that haploinsufficiency of enzymatic activity causes reduced survival or increased apoptosis of myeloid precursors because of disturbances in the hematopoietic microenvironment. Evidence against this is provided by the observation that no consistent effect of elastase mutants was found on proteolytic activity, substrate specificity or serpin inhibition,22 suggesting loss of function is not the primary mechanism responsible for neutropenia. Furthermore, elastase null mutant mice generated by gene targeting are not neutropenic.23 More recent studies suggest that elastase mutants elicit the unfolded protein response (UPR), which ultimately leads to apoptosis of granulocytic precursors. UPR is triggered by accumulation of misfolded proteins in the endoplasmic reticulum and consists of three major mechanisms. First global protein synthesis is attenuated. Second transcription of endoplasmic reticulum resident proteins involved in normal protein folding, such as the chaperone binding immunoglobulin protein (BiP), is induced. And third, endoplasmic reticulum-associated protein degradation (ERAD) of misfolded proteins is triggered. However, if the endoplasmic reticulum stress is severe, the UPR triggers apoptosis.24 Two recent studies provide evidence for a role of UPR in the pathogenesis of SCN. Several ELA2 mutants showed insufficient intracellular trafficking and accumulation of cytoplasmic elastase and cells expressing mutant elastase proteins demonstrated up-regulation of BiP and increased apoptosis, suggesting UPR activation.25 Similarly, the second study confirmed increased BiP expression in cells expressing mutant elastase, but also in granulocytic precursors isolated from patients with SCN.26
While ELA2 mutations account for about 50% of SCN cases, the original pedigree described by Kostmann3 do not carry mutations in the ELA2 gene.27 Recently three patients belonging to the original ‘Kostmann family’ were found to carry mutations in the HAX1 gene.28 HAX1 mutations are found in about a third of SCN patients and so far nine mutations have been described with the W44X mutation comprising 72% of cases.28–30 HAX1 protein is a mitochondrial molecule regulating the mitochondrial membrane potential and failure to maintain the mitochondrial membrane potential due to loss of HAX1 function result in accelerated spontaneous and induced apoptosis of neutrophils.28
A family with X-linked congenital neutropenia and monocytopenia was shown to have a mutation in the gene encoding the Wiskott–Aldrich syndrome (WAS) protein (WASp, see later).31 To date three distinct mutations in the WAS gene have been described to yield this X-linked neutropenia.31–33 These patients are quite distinct from those with WAS phenotypically, with normal or near normal platelet counts and platelet volumes, and none of the typical immunologic abnormalities. It appears that WASp is rendered constitutively active by a genetic mutation in the conserved GTPase-binding domain (based on the ability of mutant protein to enhance Arp2/3-mediated actin polymerization), and within the autoinhibitory hydrophobic core. Mutations resulting in typical or attenuated WASp almost invariably occur outside this region. The mechanism by which constitutively active WASp leads to neutropenia and monocytopenia remains unclear, but it is thought that enhanced and delocalized actin polymerization disrupts normal mitosis and leads to decreased cell proliferation and increased apoptosis of granulocyte precursor cells.34
During hematopoiesis, cellular proliferation, differentiation and survival are largely dependent on the regulation of gene expression by the actions of transcription factors. Members of the CCAAT/enhancer binding protein (C/EBP) family of transcription factors are key regulators of cellular differentiation and function in many tissues. Six homologous members of this family have been identified (C/EBPα, β, γ, δ, ε and ξ), each existing as distinct isoforms.35 All members have been shown to possess an activation domain, a DNA-binding basic region and are able to homodimerize and heterodimerize with each other through a leucine-rich dimerization domain, termed the leucine zipper, which determines specificity of dimer formation. C/EBPα, β, δ and ε are all expressed during hematopoiesis (Fig. 17.1A); however, C/EBPε, the newest member of the family, is expressed exclusively in cells of myeloid and T-cell lineages.36 Targeted disruption of the C/EBPε gene in mice results in defects of granulopoiesis, neutrophil phagocytosis and bacterial killing, migration, and impaired cytokine production after an inflammatory challenge.37,38 In addition, there is accelerated apoptosis in maturing granulocytic cells.39 Neutrophil-specific granule deficiency (SGD) is a very rare congenital disorder, possibly inherited in an autosomal recessive fashion that is characterized by a lack of specific (secondary) granule proteins such as lactoferrin, transcobalamin, collagenase, gelatinase B (tertiary granules), abnormalities of migration, disaggregation and bactericidal activity, and atypical bilobed nuclei. Primary granules are markedly depleted of defensins although expression of myeloperoxidase and lysozyme is unaffected.40 Eosinophils also lack eosinophilic-specific granules and are undetectable by Giemsa–Wright staining. Despite the lack of lactoferrin in neutrophils, saliva from patients contains normal levels suggesting a specific defect of myeloid granulopoiesis. Given the striking similarity between SGD and mutant mice generated by targeting of the C/EBPε gene it is unsurprising that mutations have now been found in at least two patients.41,42 Recently a heterozygous mutation in a third patient has been revealed, although this mutation resulted in enhanced expression of C/EBPε and another myeloid transcription factor PU.1.43 Interestingly, this SGD patient had reduced levels of the transcription factor growth factor indepence-1 (Gfi-1), which could explain the increased expression of C/EBPε and PU.1, as the expression of these are repressed by Gfi-1. Gfi-1 is a zinc-fingered transcription factor, which functions downstream of C/EBPε and PU.1, and gene targeted Gfi-1−/− mice lack mature circulating neutrophils.44 Mutations in the human Gfi-1 gene cause severe neutropenia and result in loss of repression of ELA2 and C/EBPε expression, which may impair the proliferation and survival of immature granulocytes.45,46
Most recently, mutations in the G6PC3 gene encoding the glucose-6-phosphatase were found to give rise to SCN. Linkage analysis of two families revealed a mutation in the G6PC3 gene that abolished its enzymatic activity and result in increased susceptibility to apoptosis.47 Screening of a larger cohort of patients identified seven unrelated SCN patients with mutations in the same gene and it was found that myeloid progenitor cells showed enlarged rough endoplasmatic reticulum and increased BiP levels,47 suggesting involvement of UPR as has been hypothesized for SCN caused by ELA2 mutations.
Low neutrophil counts but intact neutrophil maturation in the BM were found in four patients from one family with recurrent Streptococcus pneumoniae infection as well as hypogammaglobulinemia, short stature and hypopigmented skin.48 A mutation in the endosomal adaptor protein 14 (p14) was found to be the cause, resulting in decreased protein expression and subsequently defective lysosome function and delayed killing.48 Although this may explain their immunodeficiency, it is still unclear why these patients are neutropenic.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


