CHAPTER 32 Disorders affecting megakaryocytes and platelets
Inherited conditions
Introduction
The blood platelet is a deceptively simple cell. Though the smallest of the circulating blood elements, it derives from the largest cell in bone marrow, the megakaryocyte. Its diminutive appearance, lack of a nucleus and clear hyaloplasm (cytoplasm) made it difficult for early microscopists to recognize the platelet as a distinct entity.1–4 As a result it was the last of the circulating cellular elements to be identified.5 Anonymity, however, suited the platelet well. The cell prefers to remain nondescript, and seeks its refuge as far from the center of the column of flowing blood as possible.6 Other blood cells carry oxygen, remove carbon dioxide, supply nutrients, transport waste, and leave the circulation to participate in immune and inflammatory reactions as required, but not the platelet. It remains as quiet as possible for its 10–12 day life span. If the cell can retire in the spleen without becoming involved in any of the useful activities served by other blood elements, the platelet’s life can be considered a complete success.
Thus the platelet has no function in the circulation, except one: to keep blood flowing. It is the sentinel on guard at all times to react immediately with sites of vascular injury as soon as subendothelium is exposed. Within seconds platelets fill an injured site with a hemostatic plug that prevents further loss of blood and ultimately, with other cell systems, restores the integrity of the vascular system for normal blood flow.7–10
The platelet serves its function as the silent sentinel of the circulation very well, but, unfortunately, it does have a blind side. It does not distinguish its role in hemostasis from involvement in thrombosis. As a result participation of platelets in vaso-occlusive events leading to heart attacks, strokes or other ischemic phenomena often overshadows its value as the basic cellular unit of hemostasis.11
Structure
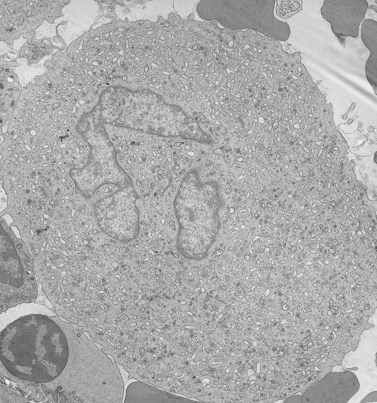
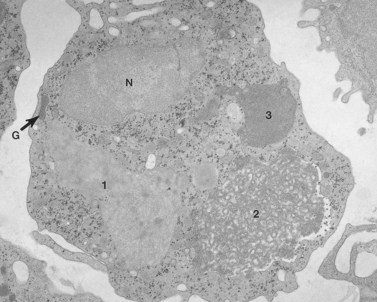
Before discussing pathology, aspects of normal megakaryocyte and platelet morphology will be considered. The megakaryocyte develops in human bone marrow from the same stem cell as do other cellular elements.12 However, its transformation from a megakaryoblast into a multinucleated giant cell is unique. The nucleus undergoes a process of endoreduplication without cell division. As a result the cell enlarges dramatically and contains many unseparated nuclear lobes (Fig. 32.1). The final number of lobes is variable, but is usually 16–32. During this process of maturation the megakaryocyte begins to form organelles including alpha granules, dense bodies and lysosomes. The surface of the giant cell invaginates into the cytoplasm forming demarcation membranes, and converts the matrix into incomplete, platelet-sized subunits. Megakaryocytes may rest at stages during maturation, but ultimately complete their development and move to endothelial cells of the bone marrow sinuses. There they extend pseudopods between endothelial cells and deliver platelets to the circulation.
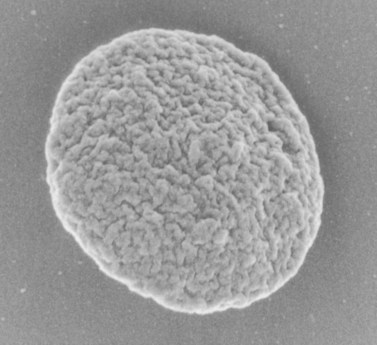
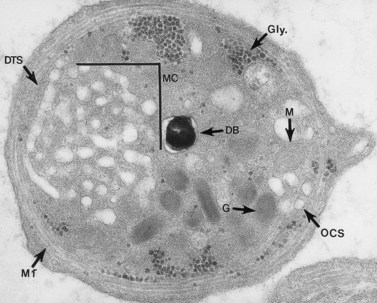
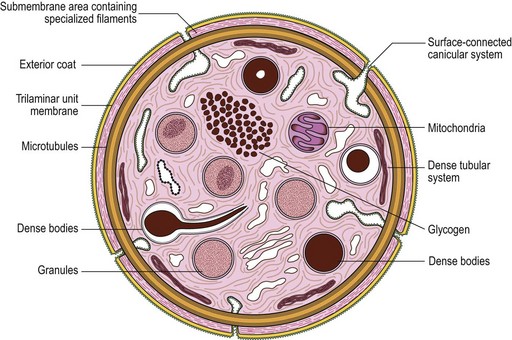
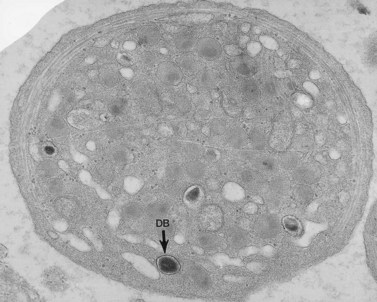
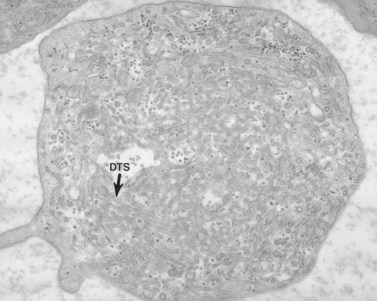
The product of this beautiful developmental sequence is rather unimpressive. It is the smallest of the cellular elements in blood and on peripheral smears resembles a speck of dirt rather than a cell. Yet, closer examination in the electron microscope reveals that the platelet is a disc similar in appearance to the discus hurled by athletes. At high magnification in the low-voltage high-resolution scanning microscope the plasma membrane is furrowed, resembling the surface of the brain13 (Fig. 32.2). Dimples appearing on the exposed surface and in replicas of freeze-fractured platelets are openings of the surface-connected open canalicular system (OCS). Thin sections in the equatorial plane reveal a circumferential coil of microtubules supporting platelet discoid form lying just below the surface membrane14 (Fig. 32.3). A large number of alpha granules, a few dense bodies and occasional lysosomes are randomly dispersed in the cytoplasm, along with mitochondria and masses of glycogen particles. Elements of the dense tubular system (DTS) of channels are also scattered randomly with two exceptions. One channel is closely associated with the circumferential coil of microtubules (Fig. 32.4). Other DTS channels are interwoven with elements of the OCS to form membrane complexes (MC). The similarity of this organization to the sarcoplasmic reticulum of embryonic muscle cells has been noted.15 Cytoplasm surrounding the organelles and other formed structures is a featureless protein matrix in thin section, but even in the resting state contains some actin filaments. Alpha granules are the most numerous of the formed organelles. They vary somewhat in size and shape, but are generally round. A nucleoid, more dense than the matrix of the alpha granule, is often seen in thin sections. Cross-sections of a few tubular elements in the matrix are von Willebrand factor concentrated in alpha granules. Dense bodies often have a typical bull’s eye appearance with the inherently opaque central core separated from the enclosing membrane by a clear space. The morphology of dense bodies, however, is extremely variable (Fig. 32.5). Some dense bodies have long, tail-like extensions or appear to be localized in alpha granules. The basis for the structural variation in dense bodies is unknown.
Disorders of megakaryocytes
In a very real sense, all of the conditions affecting the parents are visited on the progeny. Thus, except for immune thrombocytopenias, all platelet abnormalities are found in megakaryocytes. It has been easier in the past, however, to characterize the problems in platelets from circulating blood than on megakaryocytes from bone marrow. Defects in such conditions as the TAR (thrombocytopenia and absent radii) syndrome,16 therefore, remain ill defined.
Congenital megakaryocyte hypoplasia
Amegakaryocytic thrombocytopenia and congenital megakaryocyte hypoplasia are rare conditions in newborn infants.17 The cause for inability to promote conversion of stem cells into megakaryocytes is unknown, since some cases have had normal levels of thrombopoietin. Hemorrhagic complications may be mild or life-threatening. Steroids appear to be of little value, but supportive care and platelet transfusion may be successful in some cases until megakaryocyte production begins.
Fanconi anemia
Most interest has focused on the anemia in this disorder, but it should be realized that Fanconi anemia is a major cause of heritable thrombocytopenia due to megakaryocytic hypoplasia.19 It is characterized by the association of bone marrow failure and pancytopenia with other congenital anomalies affecting the musculoskeletal and genitourinary systems. While the congenital anomalies are evident at birth, the pancytopenia may be delayed for several years. Thrombocytopenia and megakaryocytic hypoplasia may be the first signs of impending bone marrow failure in Fanconi anemia.
Disorders of platelets
Platelet organelle defects
Dense bodies – general aspects
In 1951, Rand and Ried20 found that 5-hydroxytryptamine (5-HT, serotonin) was a normal constituent of platelets, and Baker et al.21 were able to demonstrate that subcellular particles separated from platelets were rich in this amine, as well as in adenosine triphosphate (ATP). Many workers subsequently confirmed the observation of Baker et al.21 and added the findings that serotonin, ATP, and ADP were located either in vacuoles or the granule fraction.22 The subcellular localization of 5-HT at the ultrastructural level, utilizing methods which had been successful in differentiating catecholamine-containing organelles in the adrenal gland, was reported by Wood.23 Employing an initial fixation in glutaraldehyde followed by exposure to potassium dichromate at low or high pH, he was able to identify organelles rich in different amines, including 5-HT. One of the cells in his report which demonstrated localization of serotonin in very dense organelles was the blood platelet. The association of serotonin with dense bodies was confirmed by ultrastructural autoradiography24 and by chemical determinations on isolated platelet subcellular organelles prepared by density gradient centrifugation.
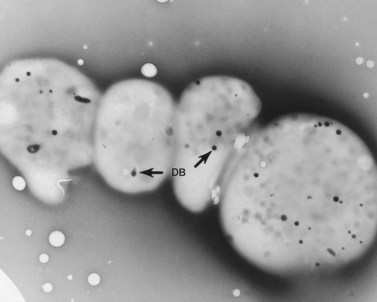
An examination of thin sections of glutaraldehyde-osmic acid-fixed platelets in our laboratory revealed a different frequency of serotonin storage organelles.25 An average of 1–1.4 dense bodies per thin sectioned platelet was found in counts on 100 cells from five normal human donors (see Fig. 32.3). Some sectioned platelets had no dense bodies in their cytoplasm. This deficiency, however, was compensated for by a significant number of cells containing 4–8 opaque organelles.
Evaluation of platelets by the whole-mount technique supported the findings made in thin-sectioned material.26 Inherently electron-opaque dense bodies were easily counted in the unstained whole mounts (see Fig. 32.5). An average of 6.15 dense bodies per platelet was found with a range of 0–24 per cell in platelets from 10 donors.
The origin of platelet dense bodies has not been specifically defined. Early work had suggested that formation of the organelles was directly related to the uptake of serotonin.23,27 Dense bodies were found only in circulating platelets, never in megakaryocytes. Later, however, it was shown that dense bodies are present in megakaryocytes from normal human bone marrows. If dense bodies are present in megakaryocytes, then some mechanism must exist for their development in the parent cell. Ultrastructural studies have suggested that such a mechanism does exist. Employing the uranaffin reaction introduced by Richards and Da Prada,28 Daimon and Gotoh29 confirmed the presence of dense bodies in megakaryocytes.
Hermansky–Pudlak syndrome (HPS)
The Hermansky–Pudlak syndrome (HPS) is a recessively inherited autosomal disease in which the triad of tyrosinase-positive oculocutaneous albinism, accumulation of ceroid-like material in reticuloendothelial cells of bone marrow (Fig. 32.6) and other tissues, and a hemorrhagic diathesis due to defective platelets are constantly associated.30 HPS occurs in patients of diverse ethnic extraction. It has been observed in American Caucasian and black populations, Argentinians, Belgians, Canadians, Czechs, Dutch, English, Finns, Germans, East Indians, Irish, Italians, Japanese, Hasidic and Ashkenazi Jews, Mexicans, Poles, Puerto Ricans, Swiss and Ukrainians.31 HPS occurs in isolates in Holland, Switzerland and Chennai, India. It is estimated that HPS occurs in approximately 1 in 2000 Puerto Ricans in the northwestern quarter of the island. The pigmentary phenotype of Puerto Rican and non-Puerto Rican HPS patients is extremely variable. Some resemble tyrosinase-negative albinos with no clinically detectable pigment in skin, hair and eyes. Most have some pigment in skin, hair and eyes and resemble tyrosinase-positive, oculocutaneous albinos. A few have deeply pigmented skin and hair, but depigmented ocular fundi, and resemble ocular albinos. However, all phenotypes include nystagmus, hypoplasia of the fovea, albinotic fundi, and decreased visual acuity.

The major cause of death in patients with HPS is fibrotic restrictive lung disease, which occurred in 43% of deceased subjects.32 All deaths from this cause occurred between 35 and 46 years of age. The second leading cause of death is hemorrhage in the perinatal period or in mothers at delivery. Sixteen per cent died from this cause. While morbidity data show that 21.6% of HPS patients have evidence of granulomatous colitis, sequelae of this condition resulted in death in only 8.1% of the deceased subjects. Thus, 67.6% died from causes directly associated with the syndrome, while 32.4% died from causes unrelated to HPS.
A granular, yellow, autofluorescent material which resembles ceroid-lipofuchsin histochemically and ultrastructurally accumulates in tissues of HPS patients. The amount of accumulation is age dependent, and the tissues in which it accumulates vary in different patients. The organs most frequently affected and in which the largest amounts of material accumulate are initially the epithelium of proximal renal tubules and later the distal tubules with little in glomeruli or the collecting tubules, bone marrow macrophages (Fig. 32.6), spleen and liver, predominantly in the portal area and in Kupfer cells. Ceroid was stored in lysosome-like structures as a granular amorphous material or occasionally with a tendency to form curvilinear or fingerprint patterns. The amount of ceroid in tissues did not always correlate with the amount of tissue damage. Tissue damage was primarily limited to gut and lung, tissues normally associated with active macrophages.
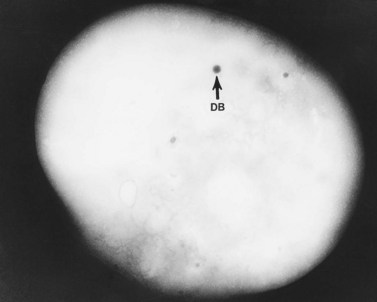
As a result of their platelet defects, HPS patients usually have a mild bleeding diathesis with ease of bruising, epistaxis and prolonged bleeding following injury, delivery or tooth extraction.30–32 Fatal hemorrhagic episodes have occurred and are often associated with the use of cyclooxygenase inhibitors such as acetylsalicylic acid. In previous reviews of ultrastructural defects in congenital disorders of platelet function it was suggested that HPS was the first disorder in which an abnormality detectable in the electron microscope could be correlated directly with a specific biochemical deficiency, impaired platelet function in vitro, and clinical bleeding problems in patients.33 The population of electron-dense bodies in HPS platelets was greatly reduced and in some cases virtually absent (Fig. 32.7). Biochemical analysis revealed that HPS platelets had very low levels of serotonin and a marked reduction in the non-metabolic pool of adenine nucleotides. However, earlier studies had shown that neither serotonin nor adenine nucleotides were responsible for the inherent opacity of platelet dense bodies, but that a concentration of heavy metal, such as calcium, impaired passage of the electron beam.26 Subsequent studies have shown that normal platelet dense bodies are rich in calcium, and that HPS platelets contain significantly less calcium than do normal cells.
Storage pool deficiency (SPD)
Patients with mild bleeding problems seemingly related to abnormal platelet secretion were recognized before the HPS was reported.34 Weiss et al.35 described six members of a family in whom secretable ADP was decreased. They postulated that their aggregation defects might be due to a specific deficiency in the non-metabolic pool of ADP. Subsequent studies of this family and other patients with a similar history and laboratory findings confirmed that defective platelet function was due to platelet storage pool deficiency (SPD). The platelet abnormality in SPD is very similar, if not identical, to that observed in HPS, even though SPD patients have normal pigmentation and no evidence of ceroid-lipofuchsin storage. As a result, it is difficult to relate the absence of dense bodies in HPS platelets to defects in melanosome formation or storage of aging pigment.
Platelets from patients with SPD are normal in size and number. Structural features of their platelets are normal, other than the marked deficiency (Fig. 32.8) or absence of dense bodies. Weiss et al.36 pointed out that the deficiency of adenine nucleotides and serotonin is less profound in SPD than in HPS platelets, and often quite variable. The bleeding in SPD patients has been found to correlate inversely with the dense body content of ATP and ADP, but appears more closely tied to the ADP level. Incubation of normal platelets with 14C-serotonin results in rapid uptake of the amine and concentration in dense bodies. In patients with SPD, the initial rate of uptake is normal, but saturation levels are decreased.36 Normal platelets retain 14C-serotonin in dense bodies for many hours, while SPD platelets lacking dense bodies rapidly lose the radioactive amine. The lost serotonin is quickly converted to 5-hydroxyindolacetic acid and 5-hydroxytryptophal by monoamine oxidases.
Platelet storage disease (SPD) has been reported to be less frequent than HPS.36 However, it may be more common. Since bleeding symptoms are mild and the pseudoalbinism and ceroid-lipofuchsin accumulation characteristic of HPS are absent, individuals with SPD may not come to the attention of physicians. Also, the degree of adenine nucleotide and serotonin deficiency in SPD platelets is variable and usually not as severe as in the HPS, resulting in further moderation of the disease. As a result, many patients with SPD may go undetected during their lifetimes.
Weiss et al.36 have provided an excellent analysis of this condition. Of 18 patients with various granule disorders, four were found to have dense body deficiency without other clinical features of HPS. In at least one family the disorder appeared to be inherited as an autosomal dominant. The hemorrhagic symptoms in SPD patients were generally mild, and they lacked the severe gastrointestinal bleeding seen in some patients with HPS. Depletion of dense body contents and electron-opaque organelles was less in SPD platelets compared to individuals with HPS. Weiss et al.36 noted that serotonin levels in SPD platelets were reduced in proportion to the reduction in platelet ADP. SPD platelets may also be deficient in their ability to synthesize intermediates of prostaglandin biosynthesis. After stimulation by collagen, SPD platelets produced less than 20% of the PGE2 and PGF2 synthesized by normal cells.
The decrease in dense bodies in SPD correlates with the deficiency in serotonin and adenine nucleotides, the impaired response of the cells to aggregating agents, and the clinical symptoms of the patient. Thus, SPD is the second disorder in which impaired platelet function can be directly associated with an ultrastructural defect in the cells. However, the normal pigmentation of individuals with this disorder and the absence of an unusual accumulation of ceroid or lipofuchsin in macrophages suggest that the cause is basically different than that responsible for platelet storage pool deficiency in HPS. The genetic basis for the SPD found in other inherited disorders, such as Wiskott–Aldrich syndrome37 and TAR syndrome38 remains to be determined.
Chediak–Higashi syndrome (CHS)
The Chediak–Higashi syndrome (CHS)39,40 is a rare, autosomally inherited disorder characterized clinically by photophobia, nystagmus, pseudoalbinism, marked susceptibility to infection, hepatosplenomegaly, lymphadenopathy and malignancy.41 Laboratory diagnosis is based on the presence of giant organelles in nearly all leukocytes on Wright-stained peripheral blood smears42 (Fig. 32.9; see also Chapter 17). The massive granules have been found in neutrophils, eosinophils, lymphocytes and monocytes from blood and in their bone marrow precursors.43
Despite the presence of thrombocytopenia, which develops during the accelerated phase of CHS, and an early report describing two patients with markedly decreased platelet serotonin,44 blood platelets have not been considered a major problem in this disease. However, several studies have shown that platelets express the genetic fault of the disorder.45–47 Platelets from patients with CHS are biochemically, physiologically and functionally abnormal.45 The defect has been related to a marked reduction in platelet-dense bodies.48,49 Elevated levels of cyclic 3′,5′-adenosine monophosphate (cAMP) were noted in platelets from one infant with CHS.45 However, the level of cAMP was corrected to normal by treatment with ascorbate without apparent improvement in platelet function.50 Thus, the platelet appears to be involved in the expression of the CHS along with other blood cells containing cytoplasmic granules.
As attractive as these findings were when first reported, there is little enthusiasm for them at present. Elevations in levels of cAMP or decreased concentrations of cGMP were not found in blood cells of other patients with CHS.51 Reversal of clinical symptoms by treatment with large amounts of vitamin C has not been found in most patients with the disease. Observations of a decrease in microtubule numbers in CHS cells52 that led to the studies of cyclic nucleotides and the suggestion to use ascorbate50 to treat the disease were not confirmed.51
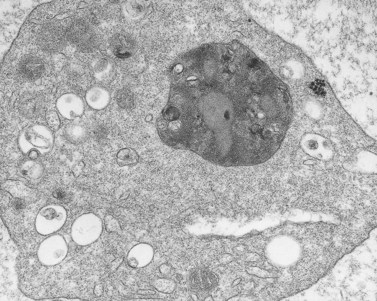
Involvement of platelets in CHS appears variable. A patient with characteristic clinical and laboratory features of the disease has been followed in this laboratory for 20 years.53 His platelets are functionally, biochemically and morphologically very close to normal (Fig. 32.10). We have studied several other patients with CHS who have storage pool deficiency, and their platelets are almost, but not completely, devoid of dense bodies.
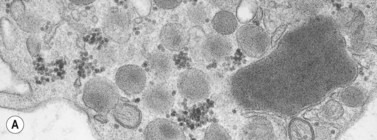
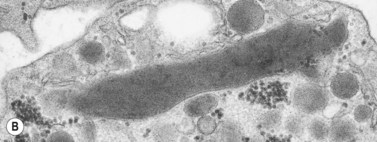
In addition to storage pool deficiency, platelets from patients with CHS have been found to contain the giant granule anomaly.46 Giant granules of a type not seen in normal platelets or in other platelet disorders were found in platelets from our patient with CHS in a ratio of about 1:100 cells in thin sections (Fig. 32.11A, B). Parmley et al.47 have confirmed this observation in another patient and have shown that the giant granules in CHS platelets are acid phosphatase positive. The relationship of the giant granule anomaly to the storage pool deficiency of CHS platelets has not been defined.
Alpha granules
Alpha granules are the most numerous of the three types of platelet storage organelles destined for secretion14,54 (see Fig. 32.3). They vary in size from 0.2 to 0.3 µm in diameter, but an occasional large granule is not uncommon in normal platelets. Alpha granules are round to oval in shape when viewed in thin sections. However, rod- and spindle-shaped alpha granules are not rare. Two zones of differing opacity are evident in the matrix of the organelle. The nucleoid is the more electron dense, and frequently has the opacity of a platelet dense body.
The lighter zone of the alpha-granule matrix usually appears unorganized. However, an occasional spindle- or rod-shaped granule can have a periodic substructure, suggesting an orderly arrangement of constituent proteins.55 Since fibrinogen is present in alpha granules, the periodicity has been related to this protein.56 Direct evidence for this, however, is lacking. Periodicity is far more apparent in whole-mount preparations than in thin sections of human and animal platelet alpha granules. For example, whole mounts of bovine platelets reveal periodicity in the substructure of every alpha granule. Tubular elements resembling microtubules in cross-section are present in alpha granules.57 Ultrastructural immunocytochemistry has shown that the tubular structures are von Willebrand factor (vWF) or that vWF is very closely associated with them.58
Biochemical studies together with evaluation of platelets from patients who lack alpha granules have provided a long list of proteins concentrated within them.59 Fibrinogen and vWF have been mentioned. Beta-thromboglobulin (β-TG), platelet factor 4 (PF-4), thrombospondin, platelet-derived growth factor (PDGF), factor V, and high-molecular-weight kininogen are also present. The list grows longer each year. Some of the alpha-granule proteins are synthesized by megakaryocytes, while others may be taken up from blood into either megakaryocytes or platelets. The ability of platelets to take up foreign particulates from plasma and transfer them to apparently intact alpha granules was reported several years ago.60 Recently, megakaryocytes have been shown to take up transfused horseradish peroxidase into alpha granules and the organelles can be detected subsequently in circulating platelets by cytochemical techniques. Recognition of this pathway is important because it appears to resolve a long-standing argument concerning the origin of platelet fibrinogen. Defibrination of animal models, followed by histochemical and cytochemical studies of bone marrow and platelets, has shown that platelet fibrinogen originates from blood.61,62
Secretion of alpha-granule contents is a characteristic feature of the platelet response to potent aggregating agents. The process of secretion has been characterized as a transfer of chemical substances confined in storage organelles of resting platelets to the exterior plasma without simultaneous loss of cytoplasmic constituents.63 Platelet release is highly selective, involving some organelles and not others, and physiologic, since it does not result from nonspecific injury.64,65
Several mechanisms have been proposed to explain how substances confined to the storage organelles in resting platelets are discharged to the exterior during the platelet release reaction. One theory suggests that organelles move to the periphery of activated platelets, fuse with the cell membrane at any point, and extrude their contents to the outside. A similar sequence of events has been observed during the process of secretion in many endocrine systems.66 However, the evidence advanced to support this mechanism in platelets is quite meager.
Ginsberg et al.67 have suggested a different mechanism for secretion of products from platelet organelles. Based on immunocytochemical and ultrastructural studies of PF4 secretion, they suggested that platelet alpha granules fuse together in the activated platelet, resulting in the formation of a large compound granule or sealed vacuole. Their evidence indicated that the sealed vacuole formed by granule fusion moves to the periphery of activated cells and fuses with the plasma membrane, resulting in release.
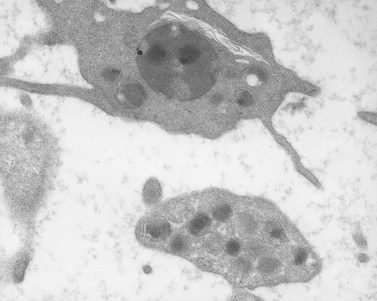
Small vacuoles do develop in thrombin-activated platelets.68 Yet the actual fusion of granules to form a compound vacuole and its movement to the cell surface as proposed by Ginsberg et al. have not been observed in activated samples. There is a swelling of the OCS and dilatation of granule membranes after communication and discharge of contents into the OCS. Granule fusion is rarely seen under these conditions, but can occur under others.69 It has been noted in platelets from patients with certain leukemias70 (Fig. 32.12), and regularly develops in platelets during long-term storage under mildly alkaline conditions71 (Fig. 32.13). Granule fusion in leukemic platelets or during storage, however, does not appear related to the release reaction.
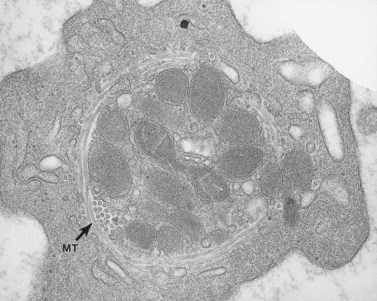
In recent studies we have examined the release reaction in bovine platelets72 and re-evaluated the secretory pathway in human cells.73 Tannic acid, often used as an electron-dense stain, was employed to delineate the process of secretion. The chemical dye was found in a preliminary investigation to precipitate fibrinogen and selectively deposit osmic acid on fibrinogen and fibrin. Samples of citrate platelet-rich plasma (C-PRP) and washed human platelets stimulated by thrombin in the presence of ethylenediaminetetraacetic acid (EDTA) develop dramatic changes in their morphology. The cells lose their lentiform appearance, become irregular in form, and extend numerous pseudopods. Platelet organelles become concentrated in cell centers and enclosed within rings of constricted microtubules (Fig. 32.14). Higher concentrations of thrombin cause rapid discharge of granule contents and reduction in their number. As a result, dense spots of actomyosin, in which centrally concentrated organelles are enclosed in less activated platelets, appear more prominent in strongly stimulated cells.74
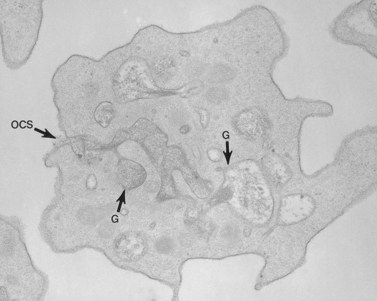
Amorphous precipitate was not present outside the thrombin-activated cells from samples of washed platelets resuspended in the presence of EDTA, and aggregates were absent. The platelets, however, revealed the same physical changes observed in thrombin-aggregated cells from platelet-rich plasma (PRP). Many granules were stained intensely by tannic acid-osmium. Other granules were swollen and their content of amorphous stained material appeared diluted. Channels of the OCS were also delineated by electron-dense stain. Some channels were tortuous and narrow and contained little tannic acid. Others were filled by electron-dense material and widely dilated. Communications between granules and OCS channels were evident in many platelets (Fig. 32.15). The connection appeared to foster swelling of the granules and dilation of the channels, so that recognition of the site of fusion was often obscured. More than one granule was frequently in communication with the same OCS channel. This relationship often resulted in extensive dilation of the OCS and granules fused to it. Occasionally, channel openings on to the surface were dilated, but usually remained constricted as in resting platelets. In some examples a single channel opened in more than one place on to the surface membrane of an activated platelet. The electron-dense material present in channels frequently appeared in the process of extrusion into the surrounding medium.
Gray platelet syndrome
The gray platelet syndrome (GPS) is a rare disorder.75 Since description of the first case, two other patients have been reported in the United States.59,76 In France, two siblings, a brother and sister, have been characterized with GPS.77 Recently, a patient from New Zealand, two in Australia, another living in England, and a family in Japan have been found to have GPS.
The original patient75 was evaluated for thrombocytopenia as a child and found to have large, nearly agranular platelets which appeared gray or blue–gray on Wright-stained blood smears. Splenectomy improved, but did not correct the platelet count to normal. It remained between 100 000 and 125 000/mm3. Most of his platelets retained the large agranular appearance noted before splenectomy, but a small percentage were of normal size and contained some granules. Since his mean platelet volume was increased (11.1 µm3), the thrombocytopenia was probably relative, as it is in other giant platelet syndromes, and the circulating platelet biomass (platelet number × mean platelet volume) was normal.78
Aggregation studies revealed an essentially normal response to most aggregating agents. However, the reaction of gray platelets to collagen and thrombin was less than normal.59 Increasing concentrations of these reagents restored the full response. Levels of serotonin and adenine nucleotides were normal. PF4, β-TG, fibrinogen, thrombomodulin and PDGF were markedly reduced. Lysosomal enzymes and catalase were within normal limits. Ultrastructural studies revealed wide variations in platelet size and morphology. Most platelets were relatively large, vacuolated, and nearly devoid of organelles (Fig. 32.16). Dense bodies, occasional mitochondria, and a few granules were present in the cells.76 Cytochemical studies with the uranaffin reaction confirmed the presence of dense bodies. A few small granules were positive for catalase and larger granules revealed reaction products for acid phosphatase and β-glucuronidase. The percentage of alpha granules was less than 15% of control platelets. Many cells were filled with elements of the dense tubular system (Fig. 32.17), while others principally contained channels of the OCS. Dilated vacuoles communicating with the OCS were common, and appeared to be sites usually occupied by alpha granules. This observation was important because megakaryocytes in patients with GPS can synthesize the proteins missing in alpha granules, but the products are lost before the large platelets reach circulating blood.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


