Chapter 21 Diseases of collagen and elastic tissue
DISEASES OF COLLAGEN: GENERALIZED DISORDERS
Ehlers-Danlos syndrome
Clinical features
The earlier Berlin classification has been replaced by that of Villefranche in which, as far as possible, the variants are subtyped by molecular mechanisms (Table 21.1).1–5 Six major types are recognized. Most show autosomal dominant inheritance, but autosomal recessive variants are also recognized.6,7 This classification is not perfect, but a more widely accepted alternative system has yet to be promulgated.
Ehlers-Danlos syndrome (EDS) is one of the most common inherited disorders, the incidence estimated being 1:5000 individuals.8,9
The clinical manifestations include:10–13
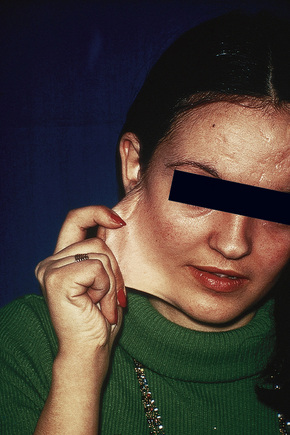
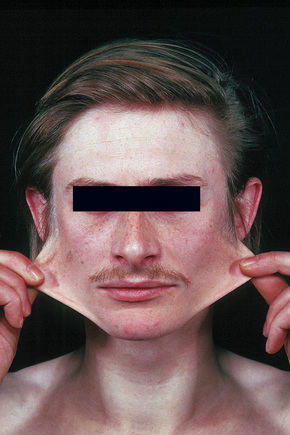
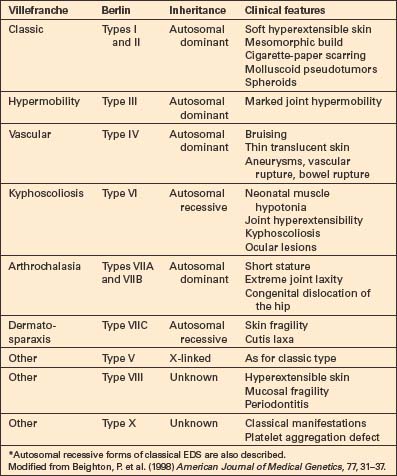
Fig. 21.3 Ehlers-Danlos syndrome: typical hyperextensible joints.
By courtesy of F.M. Pope, MD, Northwick Park Hospital, London, UK.
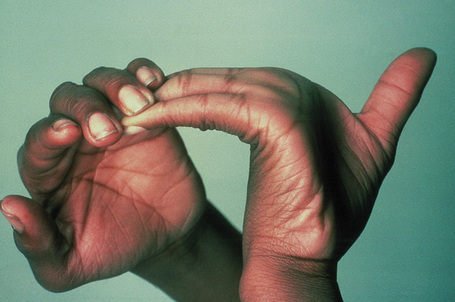
Fig. 21.4 Ehlers-Danlos syndrome: joints may be extremely lax.
By courtesy of the Institute of Dermatology, London, UK.
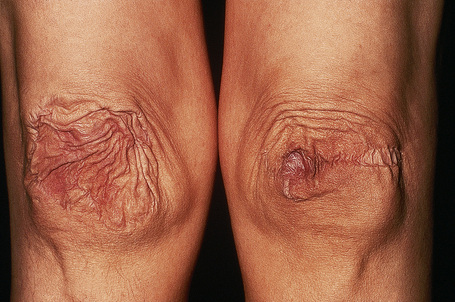
Fig. 21.5 Ehlers-Danlos syndrome: characteristic scarring involving the knees.
By courtesy of F.M. Pope, MD, Northwick Park Hospital, London, UK.
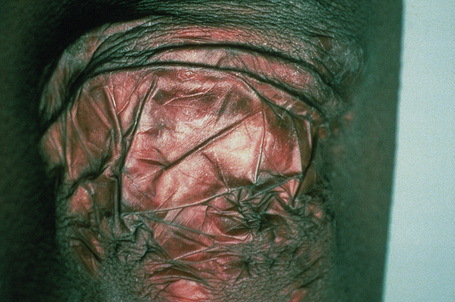
Fig. 21.6 Ehlers-Danlos syndrome: close up view of the knee of a different patient.
By courtesy of the Institute of Dermatology, London, UK.
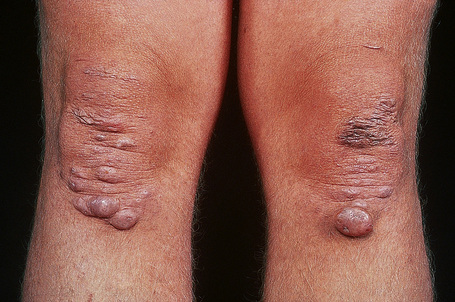
Trauma, with resultant hematoma formation and scarring, may give rise to so-called molluscoid pseudotumors (Fig. 21.7). Patients also develop hard, discrete, mobile (sometimes calcified) fibrous nodules (spheroids) in the subcutaneous fat of the extremities. Redundant skin (acquired cutis laxa) may develop over the elbows and knees, particularly in elderly patients.10,11
Gastrointestinal involvement presents as hematemesis, melena, and colonic rupture, and vascular lesions include arterial rupture and aortic or other major vessel aneurysms. Complications in pregnancy can occur.14,15 Underlying coagulopathies, dysphonia, and tongue hypermobility have been described.16–18 Mild to moderate neuromuscular symptoms including muscle weakness, myalgia, and decreased vibration sense can also be seen.19
The following briefly summarizes the salient clinical features.
Classical type EDS (Berlin types I and II)
In the Villefranche classification (Table 21.1), the classic (autosomal dominant) type includes types I (gravis) and II (mitis) in the Berlin classification, as their difference is merely one of severity of involvement. Patients suffer from soft hyperextensible skin with a tendency to split over bony eminencies.7,20 A broad mesomorphic physique with wide hands and feet is characteristic. Extensive tissue paper scarring (said to resemble a fish’s mouth) over the knees and shins is usually present and molluscoid pseudotumors are common, with the cutaneous findings more prominent than in hypermobility types.10,21 Additional features may include epicanthic folds, mitral valve prolapse, cutaneous varicosities, myopia, and late-onset osteoarthrosis.11 Complications in pregnancy include premature rupture of membranes, increasing perineal skin tearing, and prolapsing uterus and bladder.21 Breast calcification has been reported as a complication from screening mammography.22
Hypermobility type EDS (Berlin type III)
Hypermobility (autosomal dominant) type III patients have minimal cutaneous manifestations, presentation being limited to severe joint laxity with hypermobility.23 Additional uncommon findings in one study included dysphonia, Arnold-Chiari type I malformation, and dolichocolon.24 This type is difficult to diagnose given the more limited spectrum of findings; many authors believe this is identical to the benign joint hypermobility syndrome.25,26
Vascular type EDS (Berlin type IV)
Vascular EDS (type IV), autosomal dominant, is of particular importance because it is associated with the most severe vascular involvement.27–29 Patients manifest ecchymotic lesions on the knees, shins, and elbows and the features of acrogeria are often present.11,30,31 The skin is characteristically thin and translucent. Additional cutaneous manifestations include short stature, keloidal or elastotic scars, elastosis perforans serpiginosa, large eyes, lobeless ears, tooth loss, a Madonna-like facies, acro-osteolysis, diffuse scalp alopecia, and orthopedic manifestations including congenital dislocation of the hip.11,32–37 Aneurysms can arise in the aorta or other major vessels and arteriovenous fistulae and arterial dissection may be complications.11,38 Blood vessel walls are notoriously fragile, complicating surgical correction and contributing to the very high mortality associated with this variant.28,30 Bowel, pleuroperitoneal, and uterine rupture are also sometimes seen.30–32,39
Kyphoscoliosis type EDS (Berlin type VI)
Kyphoscoliosis type (type VI), autosomal recessive, presents with neonatal muscle hypotonia associated with generalized joint laxity, kyphoscoliosis, cystic malformation of the meninges, and cutaneous and ocular fragility with retinal detachment.11,40–43 This type has been further divided into type VIA which has a deficiency of lysyl hydroxylase (PLOD1 mutations) and type VIB which have normal levels of lysyl oxides.
Arthrochalasia type EDS (Berlin types VIIA and VIIB)
Arthrochalasia type (types VIIA and VIIB), autosomal dominant, presents with bilateral congenital dislocation of the hips, severe widespread hypermobility and other joint dislocations, muscular hypotonia, and hyperextensible skin with fragility.44
Dermatosparaxis type EDS (Berlin type VIIC)
Dermatosparaxis type (type VIIC), autosomal recessive, named after a condition initially described in cows and sheep, is characterized by skin fragility with tearing and joint hypermobility (Gr. sparagmos, a tearing), is extremely rare, with only a handful of cases reported.45–49 Mutations in ADAMTS2encoding procollagen I amino-peptidase are characteristic.45,47 Patients present with marked skin fragility and laxity with redundant skin folds, blue sclerae, increased bruisability, micrognathia, large fontanelles, umbilical hernia, and growth retardation.46,47,49,50
Other types of EDS (Berlin types V, VIII, X)
Pathogenesis and histological features
Ehlers-Danlos syndrome represents a heterogeneous group of genetic disorders characterized not only by abnormal fibrillar collagen synthesis but also by abnormalities in proteins that interreact with collagen. The mechanisms involved include:53
In addition, tenascin X, an extracellular matrix glycoprotein which binds to and regulates collagen fibril arrangement and possibly remodeling in the dermis, can be affected. Defects in TNXB, the gene encoding tenascin X result in abnormally clumped elastic fibers and are responsible for autosomal recessive forms of Ehlers-Danlos syndrome.54–57 The level of tenascin X may be related to neuromuscular symptoms of Ehlers-Danlos syndrome.19
The known genetic defects are summarized in Table 21.2.
Table 21.2 Molecular defects in Ehlers-Danlos syndrome
| Villefranche | Molecular defect | References |
|---|---|---|
| Classic | Mutations of COL5A1, COL5A2, TNXB (tenascin-X) | 61–64 |
| Hypermobility | Autosomal dominant, gene unknown | 24 |
| Vascular | COL3A1 mutation | 27, 32, 34, 56, 65, 66 |
| Kyphoscoliosis | Mutations in PLOD1 (lysyl hydrolase) | 40, 67, 68 |
| Arthrochalasia | Mutations of COL1A1, COL1A2 | 44, 69, 70 |
| Dermatosparaxis | Mutations of ADAMTS2(procollagen 1 amino-peptidase) | 45, 47, 71 |
Generally, histological examination of skin biopsies from patients with Ehlers-Danlos syndrome is unrewarding.58 While there may appear to be a diminution in the number and thickness of the collagen fibers, the range is so wide in normal skin that histopathological examination really has little to offer; diagnosis is essentially molecular.51
Ultrastructural observations are of limited value. In the classic variant, the resultant abnormal collagen fibrils are 25% greater in diameter, disorganized, and there are characteristic composite fibrils with a cauliflower-like appearance.10,11,50 Tenascin-X deficient fibrils are imperfectly aligned.50 In the vascular type, collagen fibrils are disorganized, irregular, fragmented, frayed, and show a variable diameter.11 Hyaluronic acid globules, amorphous matrix, microcalcifications, elastic-like fibers, and microcavities are also present.59 Similar ultrastructural findings are seen in defects in type I collagen.60 The fibrils in the arthrochalasia type are angular whereas in the dermatosparaxis type they have a hieroglyphics-like appearance.10
Aplasia cutis congenita
Clinical features
Aplasia cutis congenita (congenital absence of the skin) presents as a focal or widespread loss of skin, subcutaneous fat and sometimes deeper tissues.1–3 The incidence is approximately 3:20 000 live births.1 The vast majority involve the scalp, but the extremities and trunk can be involved.4–7 It represents a heterogeneous group of conditions of varied etiologies, which has been usefully subdivided into nine groups based on associated features, etiological factors, distribution of lesions, and patterns of inheritance.2 Usually, localized aplasia cutis is an isolated phenomenon (group 1), but in a significant proportion of patients it may signify a serious underlying congenital systemic disease.8
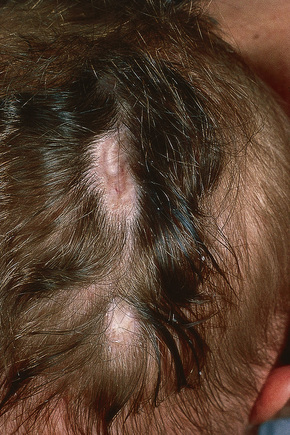
Fig. 21.8 Aplasia cutis: characteristic scarring with alopecia on the back of the head.
By courtesy of D. Atherton, MD, Institute of Dermatology and Children’s Hospital at Great Ormond Street, London, UK.
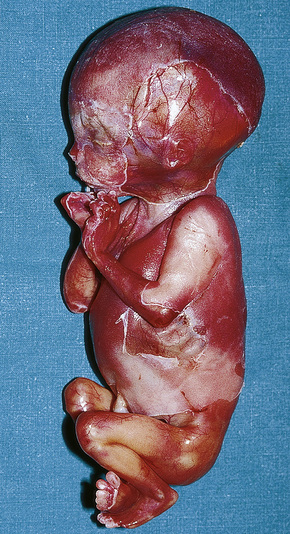
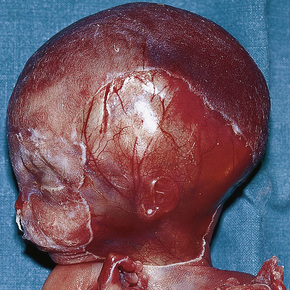
It is important that aplasia cutis is not mistaken for birth-related trauma with resultant medicolegal considerations.56 Rare, fatal cases with systemic aplasia cutis congentia have been described.57
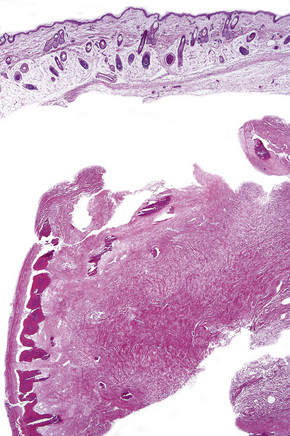
Focal dermal hypoplasia syndrome
Clinical features
Focal dermal hypoplasia (FDH; Goltz syndrome) is a rare X-link autosomal dominant disorder involving lesions of both ectoderm and mesoderm and is found almost exclusively in females.1–5 Sporadic variants are frequently encountered and a small number of affected males has been reported, possibly from postzygotic mutations.3–11 Focal dermal hypoplasia syndrome is associated with cutaneous, skeletal, ocular, oral, dental, aural, and soft tissue defects.1,2,4
Point mutations in PORCN (Xp11.23), which encodes an O-acetyltransferase that seems to facilitate secretion of ligands for the Wnt pathway, are associated with FDH.5,11–16 Sporadic cases tend to have point mutations and small deletions while familial cases show larger deletions.10 The Wnt pathway is critical for embryological development of a number of tissues including skin, fibrous, adipose, muscle, bone, teeth, nervous system, and limb patterning.17 The variability in phenotype has been attributed to mosaicism and lyonization.5,10
Cutaneous defects
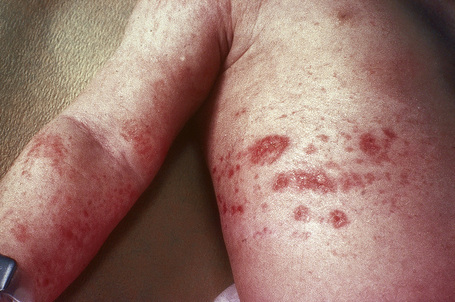
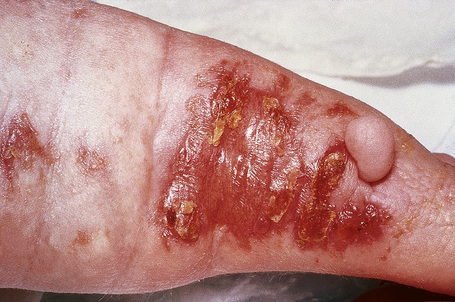
Cutaneous lesions, which are almost invariably present from birth, include areas of hypoplastic skin (such as aplasia cutis) which present clinically as depressed and atrophic cribriform and reticulated lesions, linear (Fig. 21.14) or reticular areas of erythema, hyper- or hypopigmentation, fatty tumors (most often on the limbs) (Fig. 21.15), and raspberry-like papillomatous or verrucous growths, particularly around the ostia of the body.4,18–21 Telangiectasia is a frequent manifestation.18 Linear lesions follow Blaschko’s lines and are most often seen on the limbs and cheeks.4,18,22 Nail dystrophy, anonychia, and focal alopecia can be present.4,18,23–25
Skeletal defects
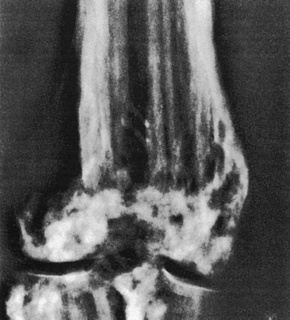
Skeletal manifestations, including hypoplasia or aplasia of the digits, ‘lobster claw hand’, syndactyly, and polydactyly, are seen in 60–70% of patients (Fig. 21.16).4,18 Facial, trunk or limb asymmetry is seen in 30%.18 Linear, vertically orientated, radiopaque striations in the metaphyses of long bones (osteopathia striata) are characteristic (Fig. 21.17).4,22 Microcrania, sometimes with mental retardation, scoliosis, kyphosis, and spina bifida occulta have also been described.18
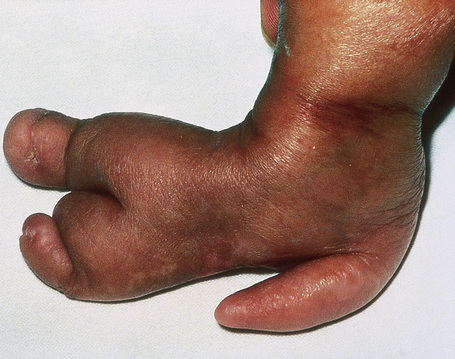
Fig. 21.16 Focal dermal hypoplasia: skeletal manifestations are an important feature.
By courtesy of D. Atherton, MD, Institute of Dermatology and Children’s Hospital at Great Ormond Street, London, UK.
Ocular, oral, dental and aural defects
Ocular findings include hypertelorism, microphthalmia, anophthalmia, aniridia, strabismus, nystagmus, glaucoma, retinal detachment, and colobomata of the iris, retina, choroid and optic nerve.20,26 Oral and dental lesions involving tooth maldevelopment (dysplasia), partial agenesis, anodontia, malocclusion, and poor enamel and papillary gingival hyperplasia may occur in up to 40% of patients.18,23,24,27–30 Aural involvement may result in both sensorineural and conduction deafness.23
Soft tissue and other defects
Soft tissue involvement can lead to striking facial asymmetry.31 The facial characteristics include prominent narrow ears, pointed chin, and nasal changes of a narrow bridge and broadened tip.18 Cleft lip and palate can occur.5,29 Genitourinary, cardiac, abdominal wall defects, and mental retardation have also been reported.13,29
Histological features
The fatty tumors show a markedly hypoplastic dermis with diminished, thinned collagen bundles and reduced elastic tissue accompanied by fatty replacement.1,3,9,32,33 Adipocytes can be clearly seen approaching or even involving the papillary dermis.4,29 Identical histological appearances are seen in nevus lipomatosus superficialis.34 Adnexa appear to be structurally normal, but may be reduced in number and situated abnormally superficial in the dermis.29 The conventional view that fatty infiltration of the dermis represents mechanical herniation has been challenged and perhaps represents a heterotopic phenomenon (i.e., an adipocytic nevus).19,28,29 The histological appearances of the verrucous and papillomatous lesions include hyperkeratosis with epidermal hyperplasia and marked papillomatosis. The dermis is thinned, with fine collagen fibers and increased vascularity.3,35 The atrophic areas show dermal thinning with sparse attenuated collagen bundles and fatty replacement.29,35 Elastic fibers may be increased and thickened.3,29 The hyperpigmented foci are characterized by acanthosis with club-shaped rete ridges and increased melanin pigmentation. Melanophages are present in the papillary dermis.29,35 By electron microscopy, collagen bundles are found loosely arranged in the extracellular matrix with smaller fibroblasts and irregular thickening of the nuclear lamina.29
Angiofibromas (adenoma sebaceum) and tuberous sclerosis
Clinical features
Tuberous sclerosis (epiloia, Bourneville’s disease) is a rare inherited systemic illness with an estimated incidence ranging broadly from 1:5800 to 1:300 000.1–3 Although an autosomal dominant mode of inheritance is sometimes seen, many if not most cases (up to 80%) appear to arise by spontaneous mutation.1–5 It comprises a constellation of cutaneous lesions as well as cerebral, ocular, cardiac, renal, pulmonary, and skeletal involvement. The sex incidence is equal and there is no racial or ethnic predilection.2
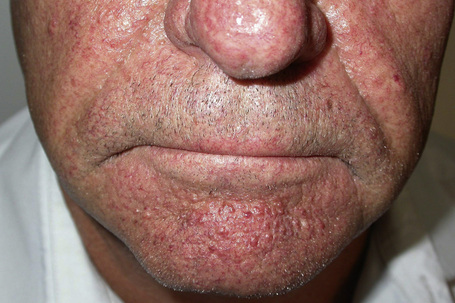
Most characteristic of tuberous sclerosis, are facial angiofibromas (adenoma sebaceum) which, while occasionally present at birth, more often appear during early childhood.6 These were initially thought to be pathognomonic, but can also be seen in multiple endocrine neoplasia type I.3,7 They are present in up to 90% of the adults with tuberous sclerosis and consist of smooth, dome-shaped, pink or reddish-brown papules and nodules (sometimes with associated telangiectases), bilaterally and symmetrically distributed, particularly in the butterfly area on the nose and cheeks and often extending to the chin, but sparing the upper lip (Fig. 21.18).3,4 Occasionally, the lesions are very extensive and develop into large cauliflower-like masses. Soft fibromas are also found about the face and scalp. Smooth, yellow or red fibrous plaques on the forehead may appear in infancy and be a presenting sign of tuberous sclerosis.3,8 Unilateral facial angiofibromas have been noted and could represent segmental expression.9 Mutiple facial angiofibromas without any known genetic disorder have been described.10
White macular lesions, which may be single or multiple, are a very frequent initial cutaneous manifestation of tuberous sclerosis.5,11–14 They are best appreciated when the skin is viewed with a Wood’s lamp3 and have been classified into three types:11
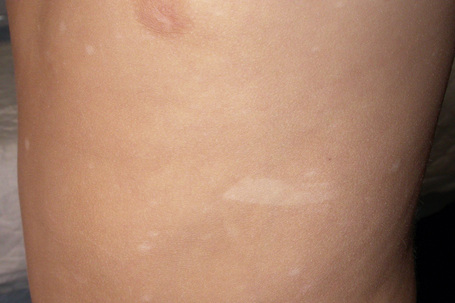
Fig. 21.19 Tuberous sclerosis: note the shagreen patch and typical ‘ash-leaf’ leukoderma.
By courtesy of J.C. Pascual, MD, Alicante, Spain.
These macular lesions are said to be present in 70% of patients with tuberous sclerosis.2 They are evident at birth or develop in the neonatal period.6 Hypopigmented patches may also affect the hair (poliosis), eyebrows, eyelashes, and occasionally the iris.5,6,8,11,12,15
Frequently seen is the shagreen patch – peau de chagrin (skin like grainy leather) – mildly yellow lesion found particularly on the lower back (see Fig. 21.19).1–7,16–18 This is a connective tissue nevus, usually presenting in early childhood or around puberty.5 Large patches in association with spina bifida occulta has been reported.19 Skin tags are often present, sometimes in large numbers.6 Other cutaneous lesions include flesh-colored sub- and periungual fibromas (Fig. 21.20).3,18 These are found in up to 50% of patients with tuberous sclerosis and usually develop at or around puberty.2,4,6 Café-au-lait macules may sometimes be present.2,3,6,7,18 Oral and eyelid angiofibromas with blepharoptosis, enamel teeth pitting, and gingival fibromas can be seen.3,20,21
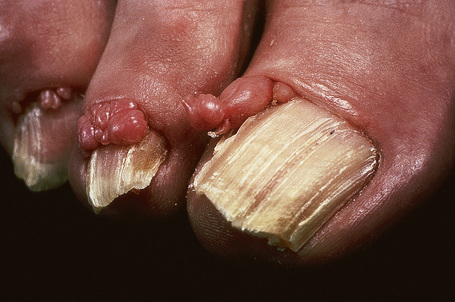
Fig. 21.20 Tuberous sclerosis: periungual fibromas are characteristic.
By courtesy of the Institute of Dermatology, London, UK.
Involvement of the central nervous system usually, but not invariably, results in mental retardation, which is sometimes severe (50–82% of patients).2,3,5,22 Epilepsy is a common manifestation (80–93% of patients) and often is the presenting symptom.2,5 Patients may develop focal or generalized tonic–clonic fits or they can suffer from infantile spasms.5 The term ‘epiloia’ was coined to define the triad of adenoma sebaceum, mental retardation, and epilepsy.7 Calcification, particularly of the basal ganglia, is radiologically detected in up to 76% of cases.23 Ophthalmological investigation reveals the presence of retinal ‘tumors’ or phakomata in up to 53% of cases, although similar lesions can be seen in neurofibromatosis.2,24 Occasionally these are calcified.
Cardiac manifestations include murmurs, electrocardiographic abnormalities, heart failure, and sudden death. Pulmonary lesions, although rare, may result in exertional dyspnea, cor pulmonale, and pneumothorax. Affected patients are usually female, often of normal intelligence, and seem less often to suffer from epilepsy.12 The prognosis is poor.
Radiological examination of the hands and feet often reveals bone cysts, focal sclerosis, and periosteal thickening.5,25
Pathogenesis and histological features
Tuberous sclerosis can result from mutations of two separate genes: TSC1 (9q34) and TSC2 (16p13).3,7,26–28 These encode the tumor suppressor proteins hamartin and tuberin, respectively. Both products interact with one another and are important regulators of cell proliferation and survival. Loss of TSC1 or TSC2 function in the mTOR pathway withdraws inhibition with numerous effects in the cells. Disregulation of Akt in the phosphatidilinositol-3-kinase (PI3K) pathway, extracellular-regulated kinase (ERK). beta-catenin, TGF-beta, and p27 have all been noted.3,7,18,28–41
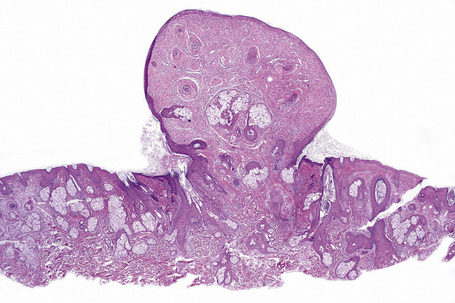
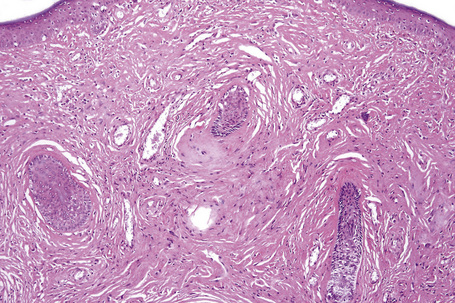
The previously applied term ‘adenoma sebaceum’ is, in fact, a misnomer, the histological features being those of a fibrovascular hamartomatous proliferation with concomitant atrophy and compression of the skin adnexae. Although often described as an angiofibroma, connective tissue nevus is a more appropriate designation. The lesion consists of an irregular proliferation of fibrous tissue and blood vessels (Figs 21.21, 21.22). Occasionally, the adnexae become surrounded and compressed by concentric lamellae of collagen (Fig. 21.23).
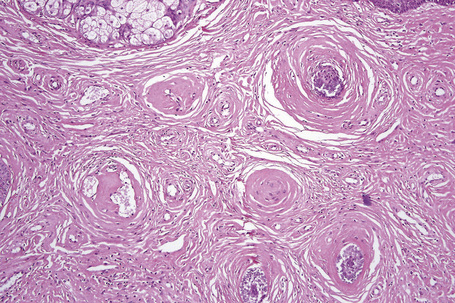
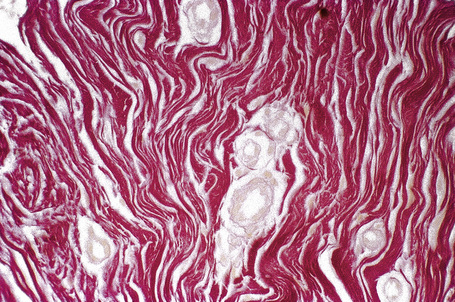
The shagreen patch is a collagenoma, the dermis being replaced by broad bundles of fibrous tissue with diminished or absent elastica (Figs 21.24, 21.25). Similarly, the sub- and periungual fibromas are fibrous tissue hamartomas.
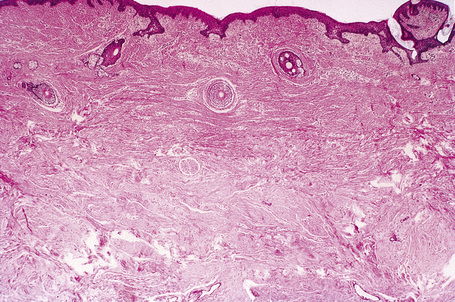
The hypopigmented macules may show normal or reduced numbers of melanocytes. Ultrastructurally, they show impaired melanogenesis (diminished numbers of small, immature melanosomes).6,11
The brain in tuberous sclerosis may be normal or diminished in size (Fig. 21.26). The neuropathology includes cortical tubers, subcortical heterotopic nodules, and subependymal giant cell astrocytomas (Figs 21.27–19.30).42,43
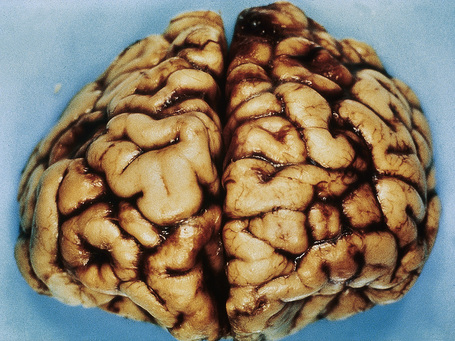
Fig. 21.26 Tuberous sclerosis: the brain is reduced in size and the gyri are markedly broadened.
By courtesy of I. Allen, MD, Royal Victoria Hospital, Belfast, UK.
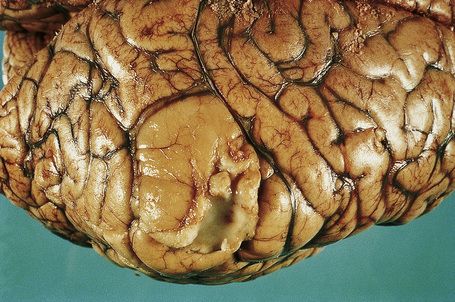
Fig. 21.27 Tuberous sclerosis: a typical ‘tuber’ on the cortex (lower midfield).
By courtesy of I. Allen, MD, Royal Victoria Hospital, Belfast, UK.
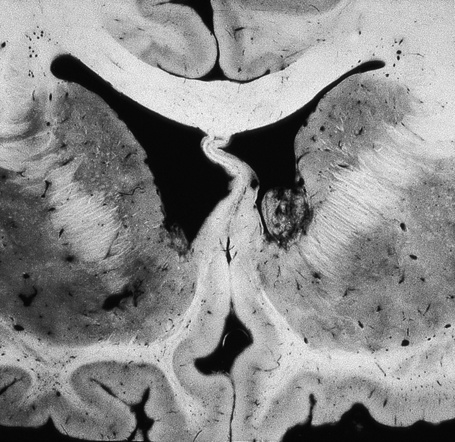
Fig. 21.28 Tuberous sclerosis: a small subependymal glial deposit projects into the right lateral ventricle.
By courtesy of I. Allen, MD, Royal Victoria Hospital, Belfast, UK.
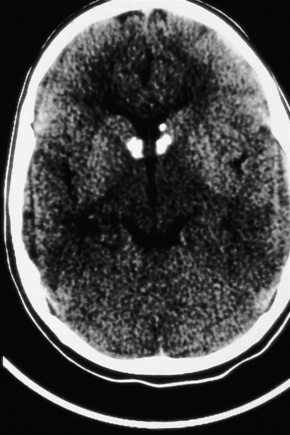
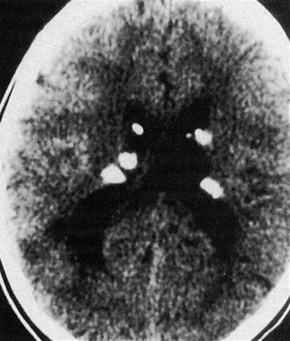
Fig. 21.30 Tuberous sclerosis: close up view.
By courtesy of the Division of Neuroradiology, University Health Center of Pittsburgh, USA.
The cardiac manifestations are due to intramyocardial rhabdomyomas, composed of large vacuolated (glycogen-filled) muscle cells.44–46 Spontaneous regression is well documented.45
The kidneys contain angiomyolipomata (often bilaterally and multiple) in up to 60% of patients and occasionally show infantile polycystic disease.5,23,47,48 Interestingly, cutaneous angiomyolipomas not associated with tuberous sclerosis are negative for MART-1 and HMB-45.49–51 Renal cell carcinoma (which may present in infancy or childhood) is also sometimes encountered.52,53 A few patients with tuberous sclerosis complex also have mutations in the nearby PKD1 gene, resulting in autosomal dominant polycystic kidney disease.28,54
Multiple cysts (lymphangiomyomatosis) are the most important pulmonary manifestation.3,55–58 Females are predominantly affected.57 Angiomyolipoma at this site is exceptional.59
There may be an association with neuroendocrine (parathyroid, pituitary, and pancreatic) tumors.60–62
Nephrogenic systemic fibrosis
Clinical features
Nephrogenic systemic fibrosis (formerly known as nephrogenic fibrosing dermopathy) is a multiorgan disease that can have a scleroderma-like dermatosis and is associated with renal failure and exposure to gadolinium.1–22
Although it was initially thought to develop only in individuals undergoing hemodialysis, subsequent publications documenting its occurrence in patients being treated with peritoneal dialysis, or following transplantation or solely with renal failure, broadened the definition.1–3,23–26 Most patients have chronic kidney disease with the vast majority having stage 5 disease (estimated GFR < 15 mL per minute per 1.73 m2). Disease course can parallel renal function.27 Some patients with acute renal disease have also been reported to develop this condition.28–31 Although the vast majority of documented patients are adults, presentation in children with renal insufficiency has been seen.32–36 While several large studies have demonstrated an association with gadolinium exposure and renal failure, clearly not all patients with these conditions develop nephrogenic sclerosis fibrosis. 20,37–45 Some of this variability may be due to the type of gadolinium contrast used.46 Additional reported risk factors include cumulative exposure to gadolinium, erythropoietin exposure, and elevated ionized calcium and phosphate serum concentrations.47
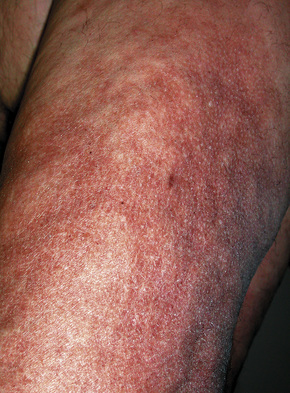
Although previously considered primarily a cutaneous disease, it is now known to affect other organs, as documented in several autopsy cases.48–52 Extracutaneous findings can be seen, including in the deep soft tissue, thyroid, heart, kidney, dura, and lung.48–53 In the skin, the condition is characterized by rapidly growing, symmetrical skin-colored or erythematous papules and nodules that coalesce to form erythematous or hyperpigmented progressive, brawny plaques, sometimes with a peau d’orange appearance (Figs 21.31, 21.32).3,5,12,28,54,55 These lesions on the breast sometimes mimic inflammatory breast cancer.56 The plaques have an irregular border and islands of sparing are sometimes described.26,55 The limbs and trunk are predominantly affected. The face is usually uninvolved although there are a number of cases with yellow scleral plaques.24,32,55 The skin is markedly thickened, hard, and has a wooden texture.3,55 Lesions may be pruritic, painful or associated with a burning sensation.26 Flexion contractures are a common complication, often resulting in considerable immobility.3 As a consequence, morbidity is high.5 Occasionally, patients may develop redundant skin similar to cutis laxa and generalized elastolysis.57,58 Mortality can occur due to thromboembolism.48
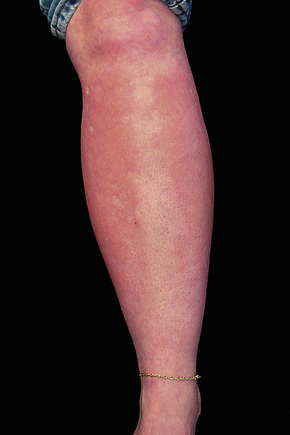
Fig. 21.31 Nephrogenic fibrosing nephropathy: note the swelling and erythema.
By courtesy of S.E. Cowper, MD, Yale University, New Haven, USA.
Pathogenesis and histological features
The exact mechanism of nephrogenic systemic fibrosis is still being unraveled. The presence of high levels of gadolinium due to renal failure is thought to result in transmetallation, where various serum ions can interact with the gadolinium contrast complex, releasing free gadolinium.22,59 Free gadolinium is toxic in animal studies and can be detected in tissue specimens from patients with nephrogenic systemic fibrosis.4,16–18 In vitro studies have shown gadolinium to be profibrotic and proinflammatory, eliciting growth factors from monocytes.60–63
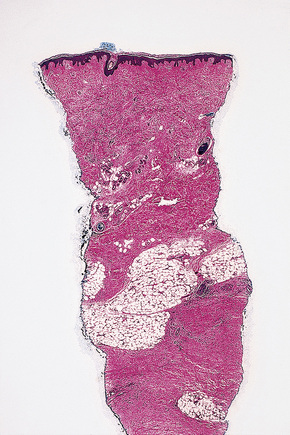
Histologically, early lesions are characterized by a subtle proliferation of fibroblasts and variable numbers of large epithelioid and stellate cells (Figs 21.33–21.35).2,64 In an established plaque, there are conspicuous CD34+ dermal dendritic cells with admixed CD68+ macrophages and factor XIIIa-positive cells (Figs 21.36–21.38).2,32,64 Multinucleate forms are present and can show an osteoclast-like morphology.65,66 There are numerous fibroblasts associated with irregularly distributed thickened collagen bundles and increased numbers of elastic fibers. Sclerotic bodies or elastocollagenous balls, irregularly shaped paucicellular areas of collagen which can ossify, have been described.67,68 Cleftlike spaces are typically present and there is increased dermal stromal mucin. Progression of the lesion is accompanied by capillary proliferation. During the earlier stages of the illness myofibroblasts have also been identified but they are typically absent in more chronic lesions. Osseous metaplasia and dystrophic calcification is sometimes evident.2,24,64,65,69–71 Calciphylaxis has been documented in several cases.70,72,73
< div class='tao-gold-member'>
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


